

Functional cloning of recurrence-specific antigens identifies molecular targets to treat tumor relapse
- PMID: 23752316
- PMCID: PMC3734666
- DOI: 10.1038/mt.2013.116
Functional cloning of recurrence-specific antigens identifies molecular targets to treat tumor relapse
Abstract
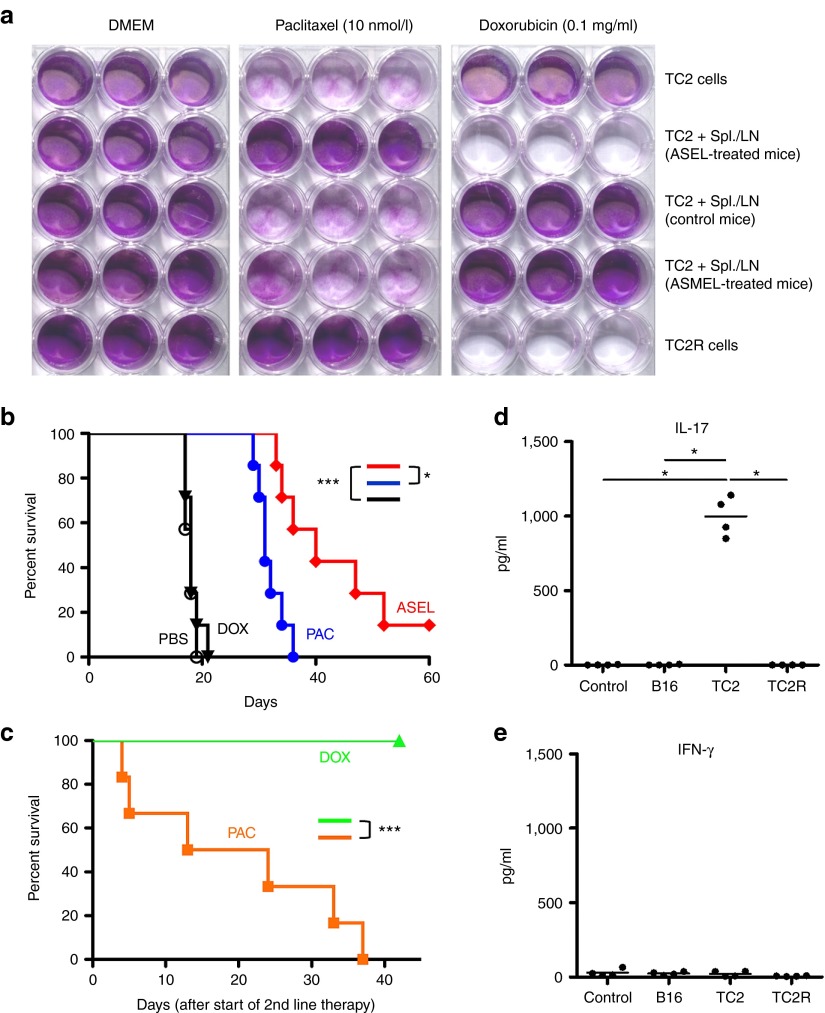
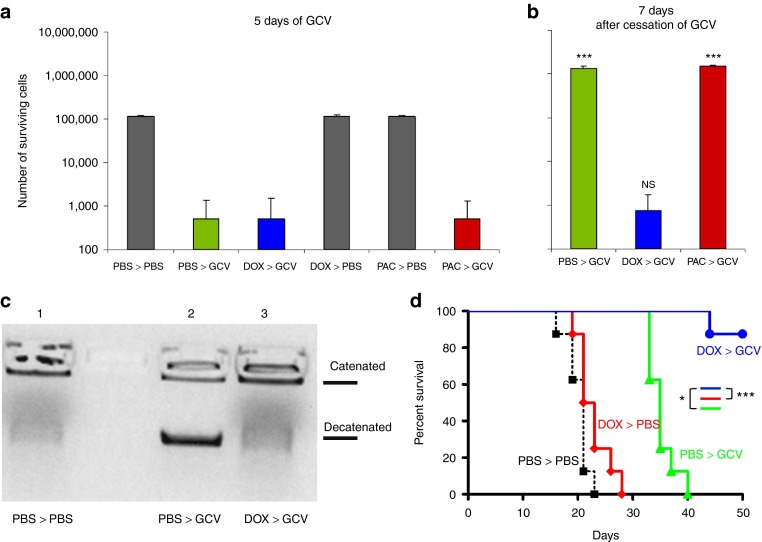
Aggressive regrowth of recurrent tumors following treatment-induced dormancy represents a major clinical challenge for treatment of malignant disease. We reported previously that recurrent prostate tumors, which underwent complete macroscopic regression followed by aggressive regrowth, could be cured with a vesicular stomatitis virus (VSV)-expressed cDNA library derived from recurrent tumor cells. By screening the protective, recurrence-derived VSV-cDNA library, here we identify topoisomerase-IIα (TOPO-IIα) as a recurrence-specific tumor antigen against which tolerance can be broken. Tumor recurrences, in two different types of tumor (prostate and melanoma), which had evaded two different frontline treatments (immunotherapy or chemotherapy), significantly overexpressed TOPO-IIα compared with their primary tumor counterparts, which conferred a novel sensitivity to doxorubicin (DOX) chemotherapy upon the recurrent tumors. This was exploited in vivo using combination therapies to cure mice, which would otherwise have relapsed, after suboptimal primary therapy in both models. Our data show that recurrent tumors-across histologies and primary treatments-express distinct antigens compared with the primary tumor which can be identified using the VSV-cDNA library technology. These results suggest that it may be possible to design a few common second-line therapies against a variety of tumor recurrences, in some cases using agents with no obvious activity against the primary tumor.
Figures
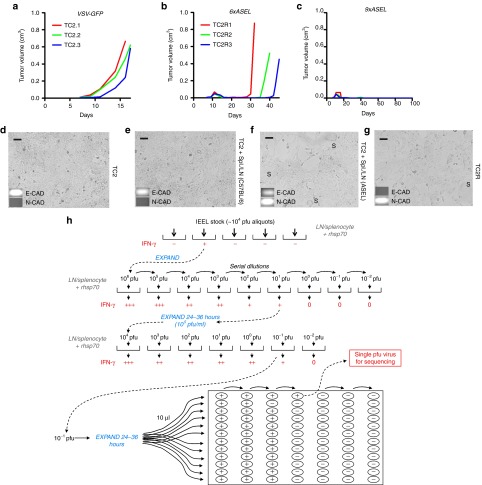
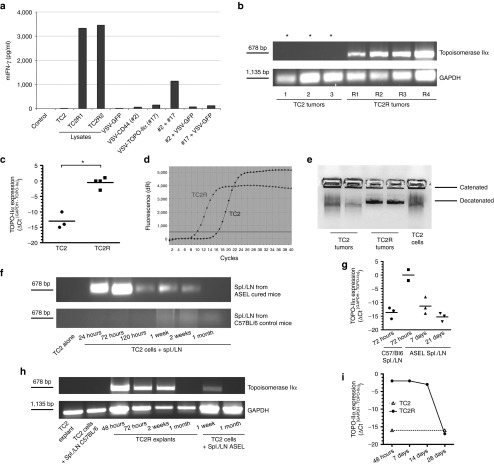
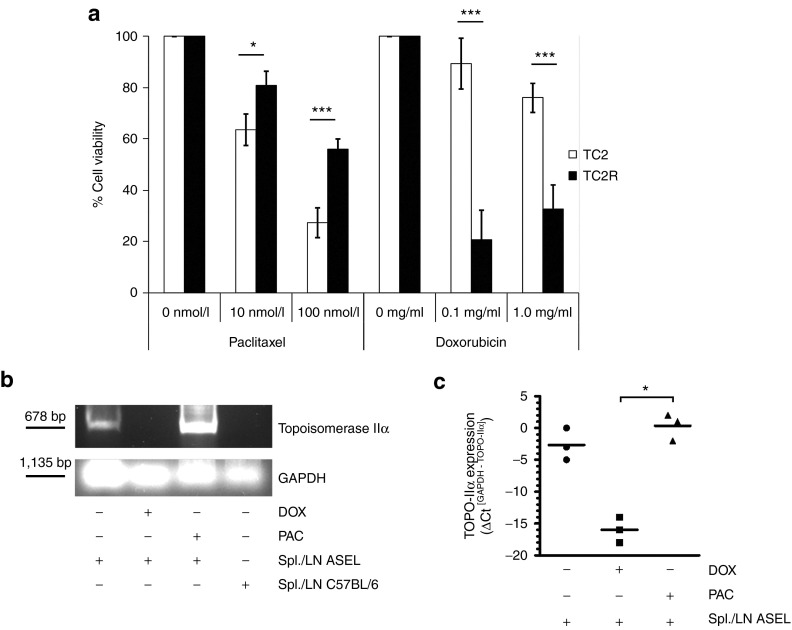
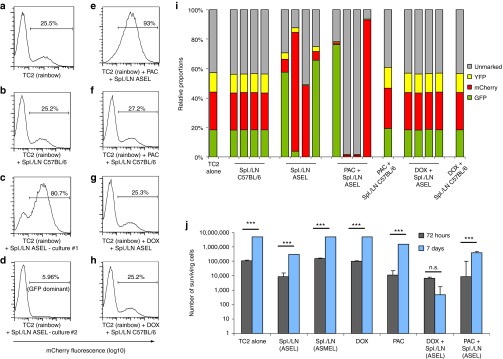
Comment in
-
Immunotherapy exposes cancer stem cell resistance and a new synthetic lethality.Mol Ther. 2013 Aug;21(8):1472-4. doi: 10.1038/mt.2013.160. Mol Ther. 2013. PMID: 23903573 Free PMC article. No abstract available.
References
-
- Goss PE, Chambers AF. Does tumour dormancy offer a therapeutic target. Nat Rev Cancer. 2010;10:871–877. - PubMed
-
- Hensel JA, Flaig TW, Theodorescu D. Clinical opportunities and challenges in targeting tumour dormancy. Nat Rev Clin Oncol. 2013;10:41–51. - PubMed
-
- McGowan PM, Kirstein JM, Chambers AF. Micrometastatic disease and metastatic outgrowth: clinical issues and experimental approaches. Future Oncol. 2009;5:1083–1098. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
