

Long QT syndrome: beyond the causal mutation
- PMID: 23753525
- PMCID: PMC3779107
- DOI: 10.1113/jphysiol.2013.254920
Long QT syndrome: beyond the causal mutation
Abstract
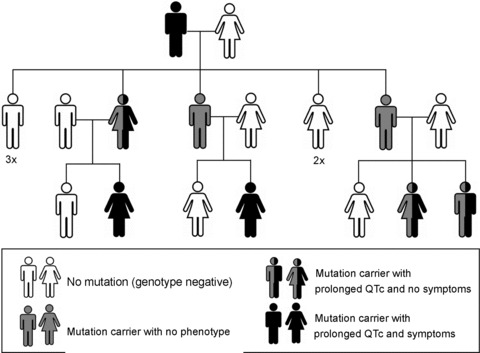
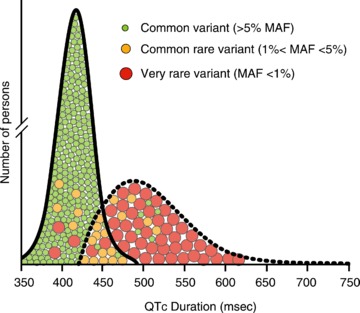
Congenital long QT syndrome (LQTS) is caused by single autosomal-dominant mutations in a gene encoding for a cardiac ion channel or an accessory ion channel subunit. These single mutations can cause life-threatening arrhythmias and sudden death in heterozygous mutation carriers. This recognition has been the basis for world-wide staggering numbers of subjects and families counselled for LQTS and treated based on finding (putative) disease-causing mutations. However, prophylactic treatment of patients is greatly hampered by the growing awareness that simple carriership of a mutation often fails to predict clinical outcome: many carriers never develop clinically relevant disease while others are severely affected at a young age. It is still largely elusive what determines this large variability in disease severity, where even within one pedigree, an identical mutation can cause life-threatening arrhythmias in some carriers while in other carriers no disease becomes clinically manifested. This suggests that additional factors modify the clinical manifestations of a particular disease-causing mutation. In this article, potential demographic, environmental and genetic factors are reviewed, which, in conjunction with a mutation, may modify the phenotype in LQTS, and thereby determine, at least partially, the large variability in disease severity.
Figures

 , stimulatory effect;
, stimulatory effect;  , inhibitory effect; 5′UTR, 5′ untranslated region; 3′UTR, 3′ untranslated region; M2-R, M2 muscarinic acetylcholine receptor; β-AR, β-adrenergic receptor; Nav1.5, INa channel protein; Kv11.1, IKr channel protein; Kv7.1, IKs channel protein; TNF, tumour necrosis factor; IL-1, interleukin-1.
, inhibitory effect; 5′UTR, 5′ untranslated region; 3′UTR, 3′ untranslated region; M2-R, M2 muscarinic acetylcholine receptor; β-AR, β-adrenergic receptor; Nav1.5, INa channel protein; Kv11.1, IKr channel protein; Kv7.1, IKs channel protein; TNF, tumour necrosis factor; IL-1, interleukin-1.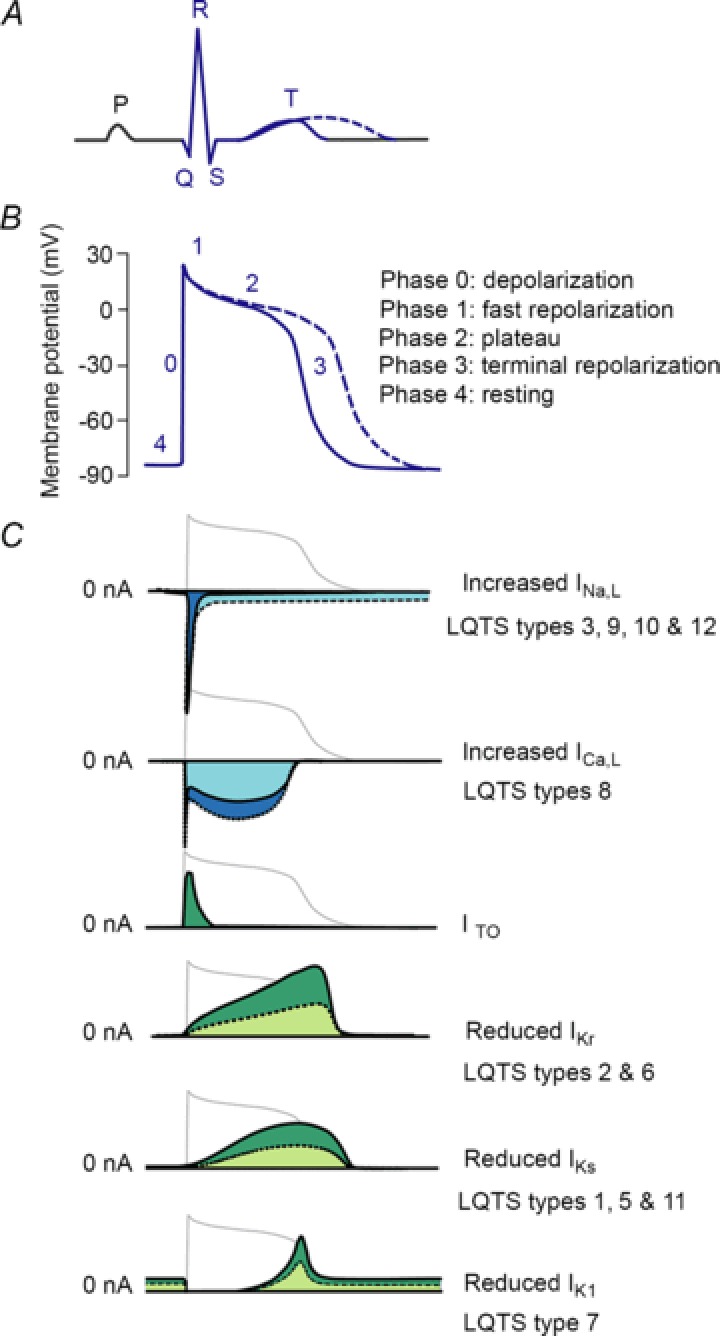
References
-
- Abbott GW, Sesti F, Splawski I, Buck ME, Lehmann MH, Timothy KW, Keating MT, Goldstein SA. MiRP1 forms IKr potassium channels with HERG and is associated with cardiac arrhythmia. Cell. 1999;97:175–187. - PubMed
-
- Algra A, Tijssen JG, Roelandt JR, Pool J, Lubsen J. QTc prolongation measured by standard 12-lead electrocardiography is an independent risk factor for sudden death due to cardiac arrest. Circulation. 1991;83:1888–1894. - PubMed
-
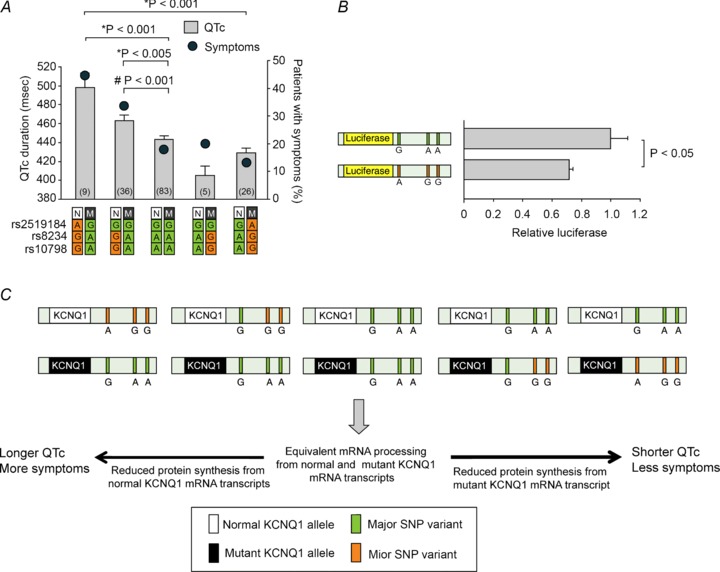
- Amin AS, Giudicessi JR, Tijsen AJ, Spanjaart AM, Reckman YJ, Klemens CA, Tanck MW, Kapplinger JD, Hofman N, Sinner MF, Müller M, Wijnen WJ, Tan HL, Bezzina CR, Creemers EE, Wilde AA, Ackerman MJ, Pinto YM. Variants in the 3′ untranslated region of the KCNQ1-encoded Kv7.1 potassium channel modify disease severity in patients with type 1 long QT syndrome in an allele-specific manner. Eur Heart J. 2012;33:714–723. - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources