

Epigenetic functions of smchd1 repress gene clusters on the inactive X chromosome and on autosomes
- PMID: 23754746
- PMCID: PMC3753908
- DOI: 10.1128/MCB.00145-13
Epigenetic functions of smchd1 repress gene clusters on the inactive X chromosome and on autosomes
Abstract
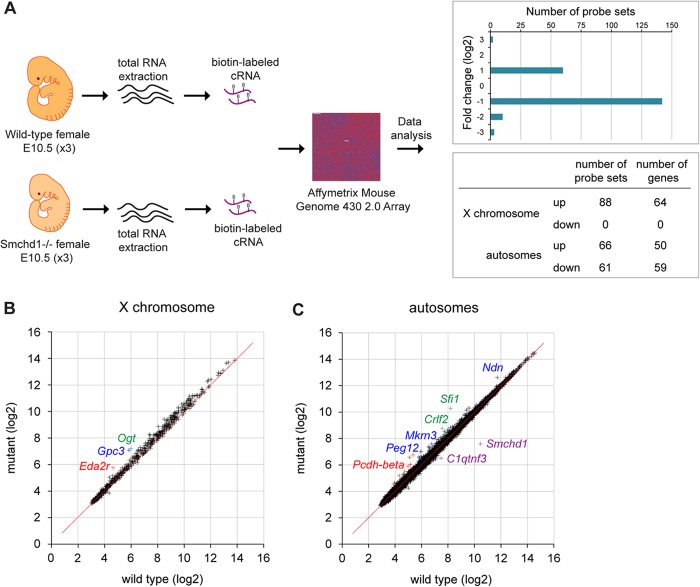
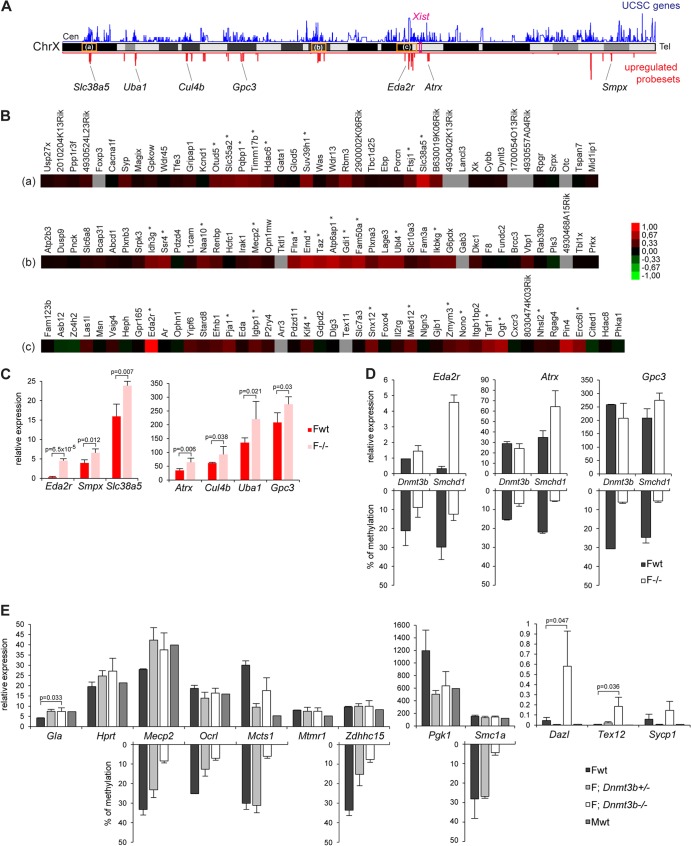
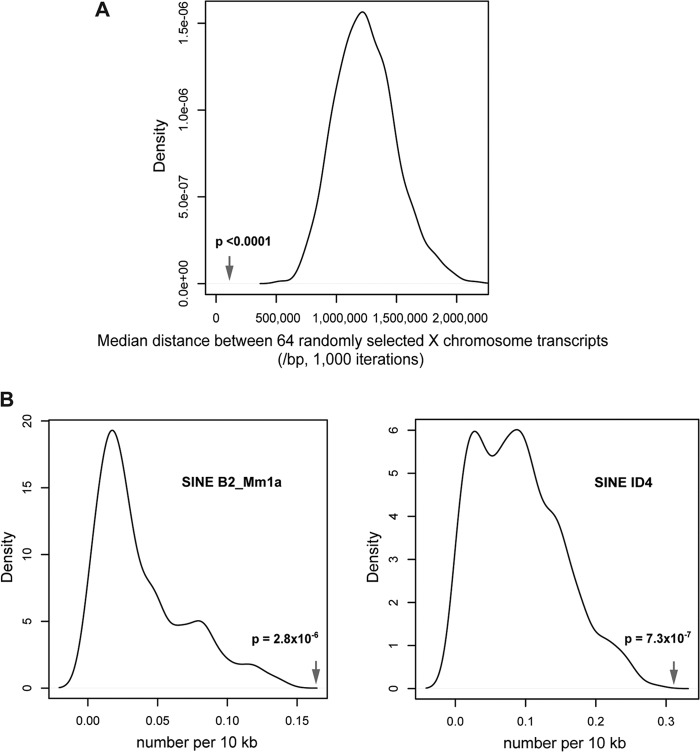
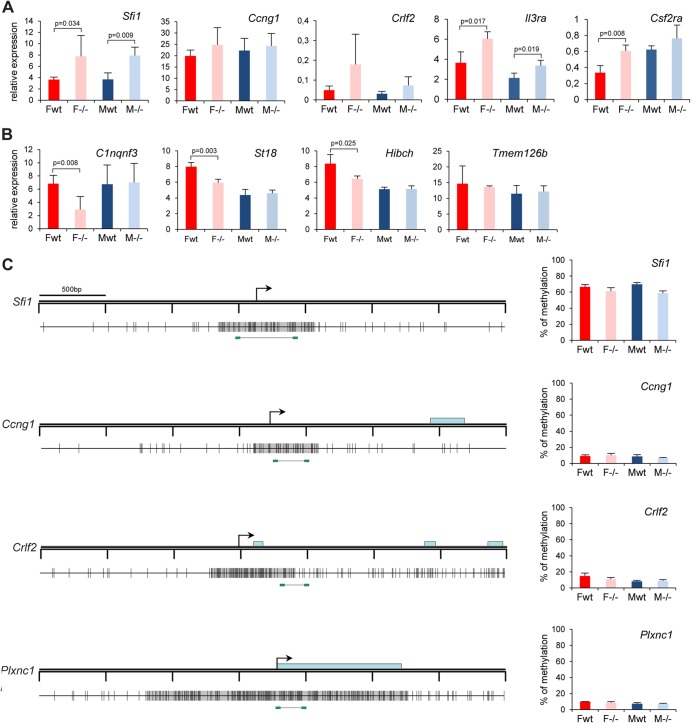
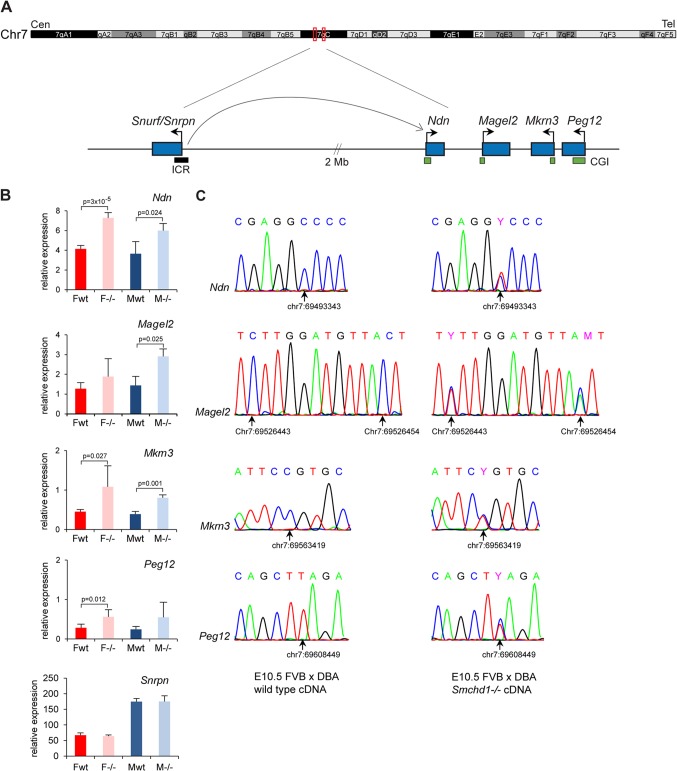
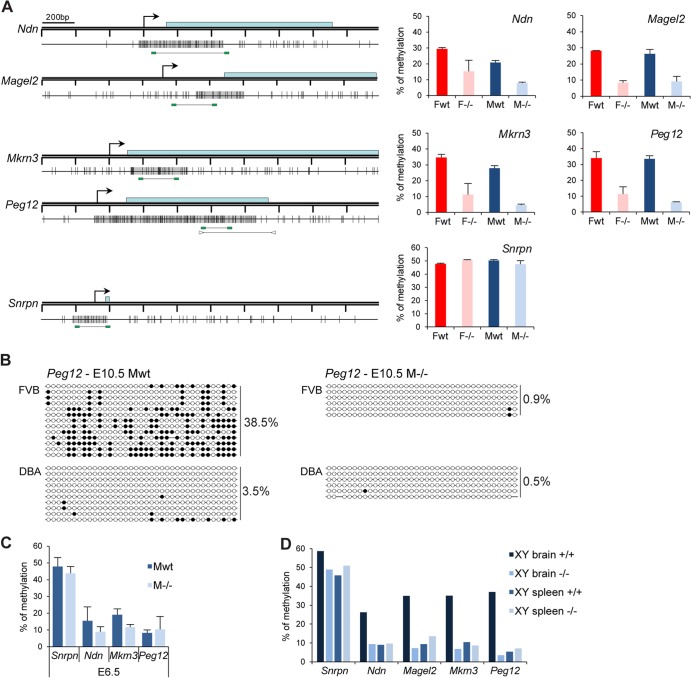
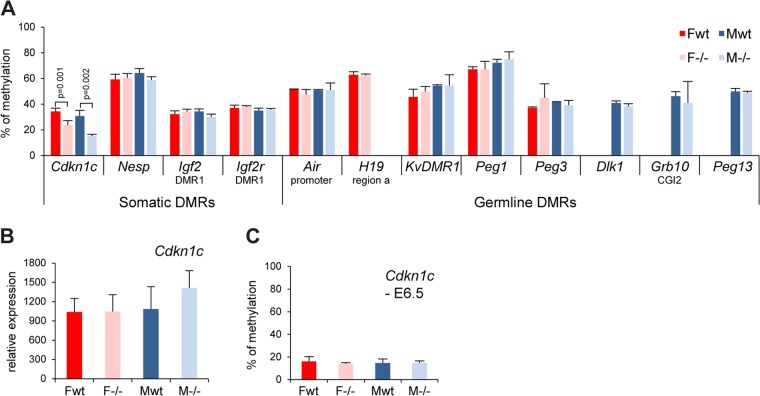
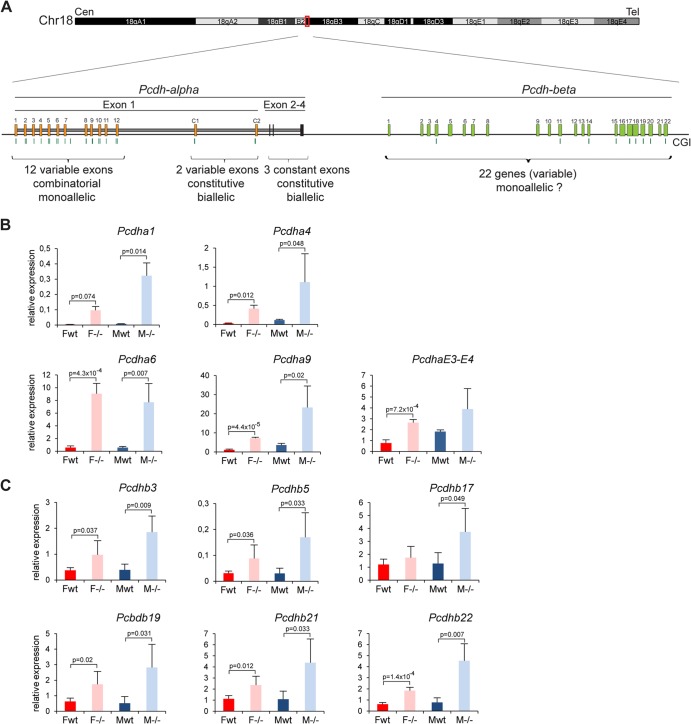
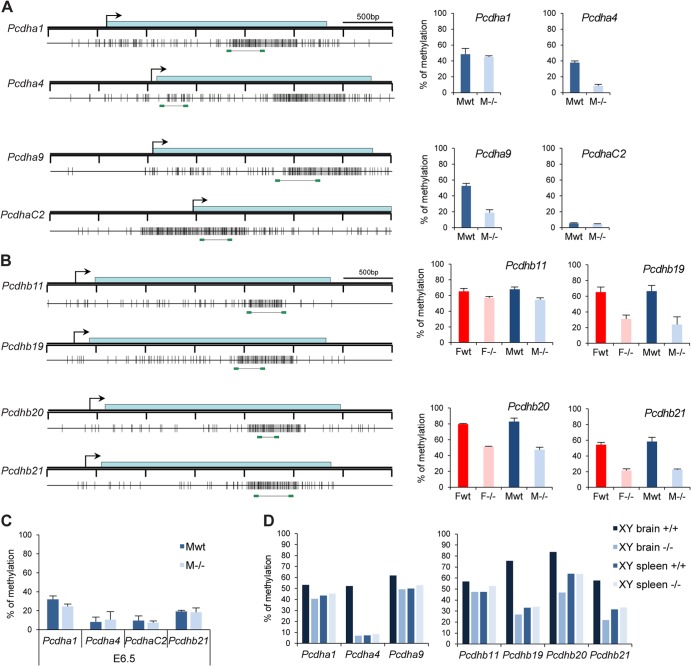
The Smchd1 gene encodes a large protein with homology to the SMC family of proteins involved in chromosome condensation and cohesion. Previous studies have found that Smchd1 has an important role in CpG island (CGI) methylation on the inactive X chromosome (Xi) and in stable silencing of some Xi genes. In this study, using genome-wide expression analysis, we showed that Smchd1 is required for the silencing of around 10% of the genes on Xi, apparently independent of CGI hypomethylation, and, moreover, that these genes nonrandomly occur in clusters. Additionally, we found that Smchd1 is required for CpG island methylation and silencing at a cluster of four imprinted genes in the Prader-Willi syndrome (PWS) locus on chromosome 7 and genes from the protocadherin-alpha and -beta clusters. All of the affected autosomal loci display developmentally regulated brain-specific methylation patterns which are lost in Smchd1 homozygous mutants. We discuss the implications of these findings for understanding the function of Smchd1 in epigenetic regulation of gene expression.
Figures
References
-
- Hirano T. 2006. At the heart of the chromosome: SMC proteins in action. Nat. Rev. Mol. Cell Biol. 7:311–322 - PubMed
-
- Blewitt ME, Gendrel AV, Pang Z, Sparrow DB, Whitelaw N, Craig JM, Apedaile A, Hilton DJ, Dunwoodie SL, Brockdorff N, Kay GF, Whitelaw E. 2008. SmcHD1, containing a structural-maintenance-of-chromosomes hinge domain, has a critical role in X inactivation. Nat. Genet. 40:663–669 - PubMed
-
- Gendrel AV, Apedaile A, Coker H, Termanis A, Zvetkova I, Godwin J, Tang YA, Huntley D, Montana G, Taylor S, Giannoulatou E, Heard E, Stancheva I, Brockdorff N. 2012. Smchd1-dependent and -independent pathways determine developmental dynamics of CpG island methylation on the inactive X chromosome. Dev. Cell 23:265–279 - PMC - PubMed
-
- Kanno T, Bucher E, Daxinger L, Huettel B, Bohmdorfer G, Gregor W, Kreil DP, Matzke M, Matzke AJ. 2008. A structural-maintenance-of-chromosomes hinge domain-containing protein is required for RNA-directed DNA methylation. Nat. Genet. 40:670–675 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases