

Benchmarking short sequence mapping tools
- PMID: 23758764
- PMCID: PMC3694458
- DOI: 10.1186/1471-2105-14-184
Benchmarking short sequence mapping tools
Abstract
Background: The development of next-generation sequencing instruments has led to the generation of millions of short sequences in a single run. The process of aligning these reads to a reference genome is time consuming and demands the development of fast and accurate alignment tools. However, the current proposed tools make different compromises between the accuracy and the speed of mapping. Moreover, many important aspects are overlooked while comparing the performance of a newly developed tool to the state of the art. Therefore, there is a need for an objective evaluation method that covers all the aspects. In this work, we introduce a benchmarking suite to extensively analyze sequencing tools with respect to various aspects and provide an objective comparison.
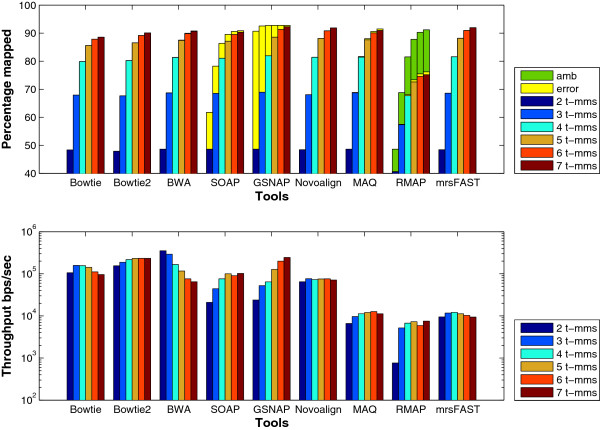
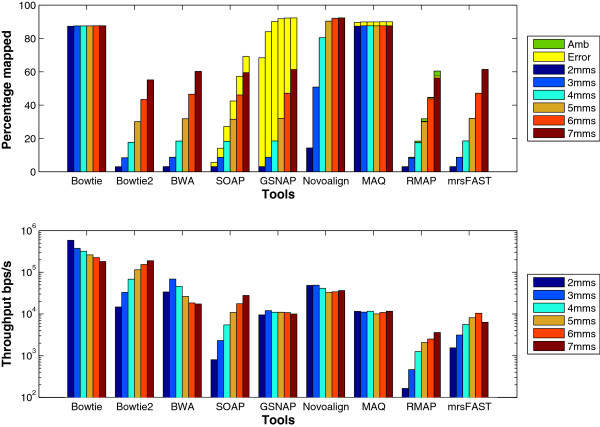
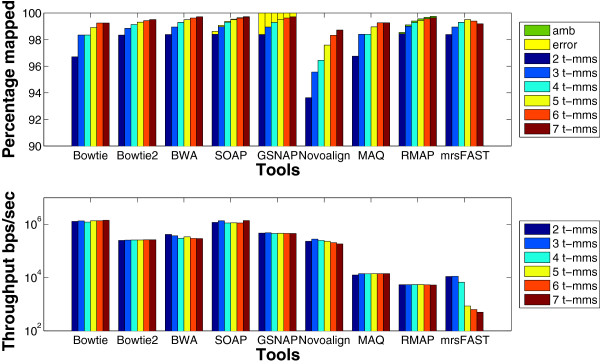
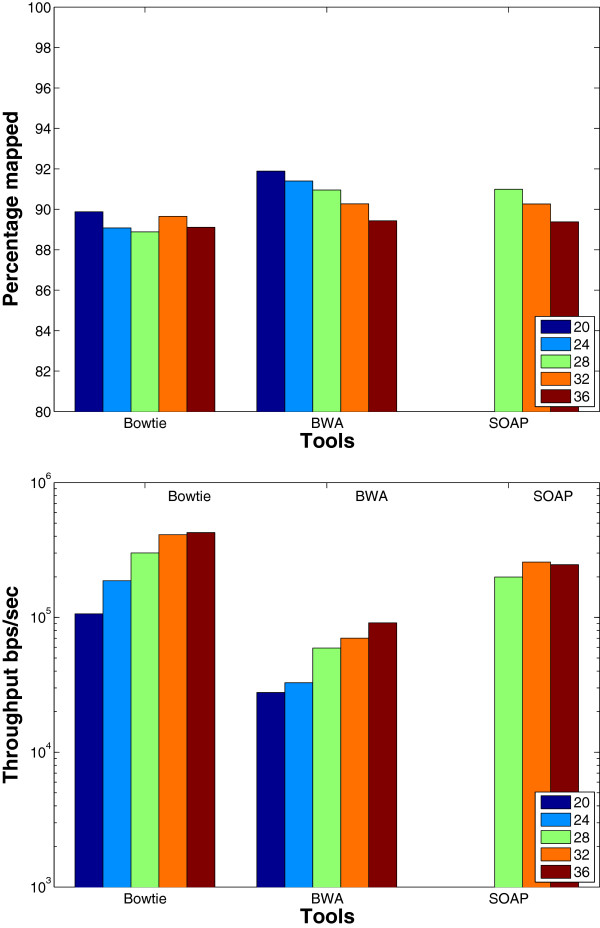
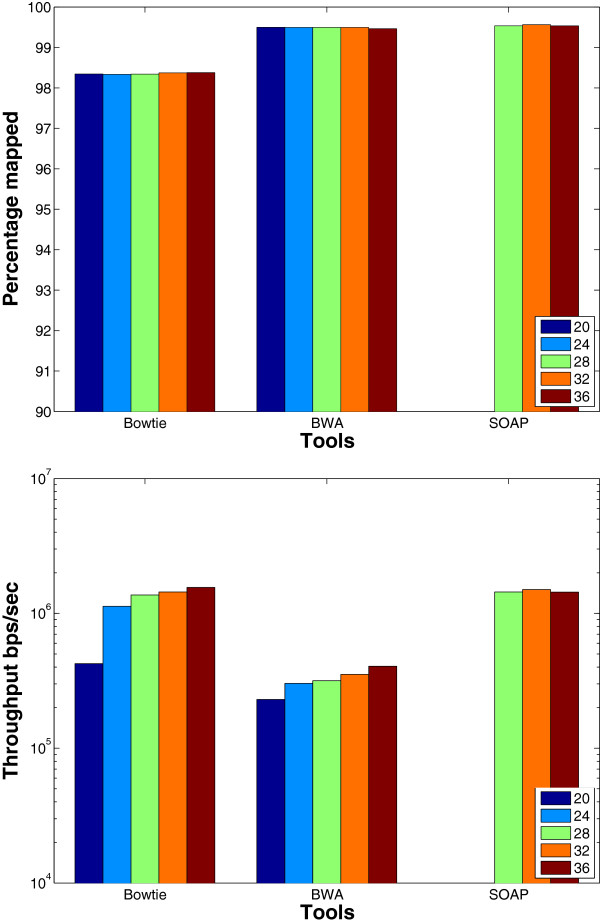
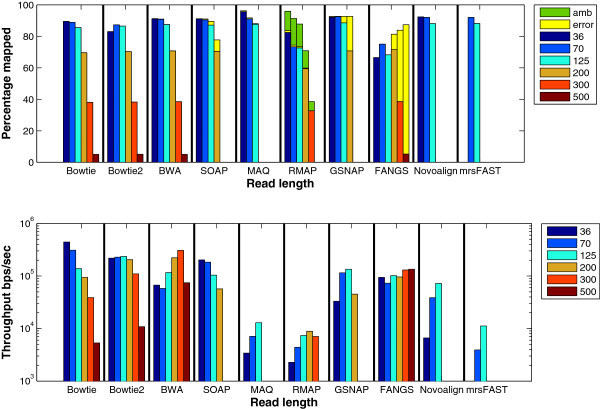
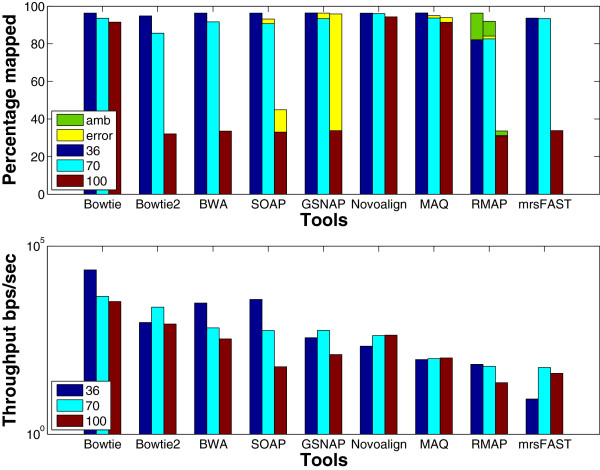
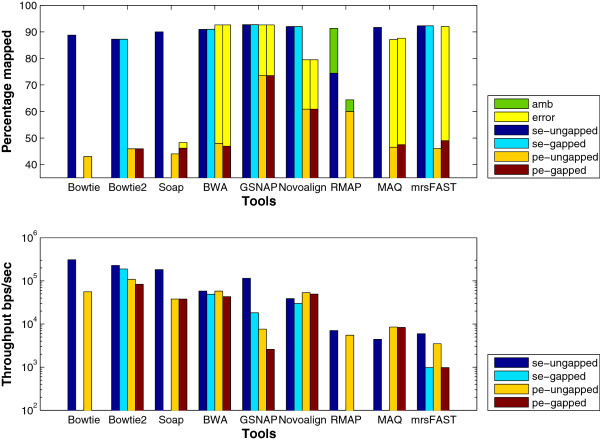
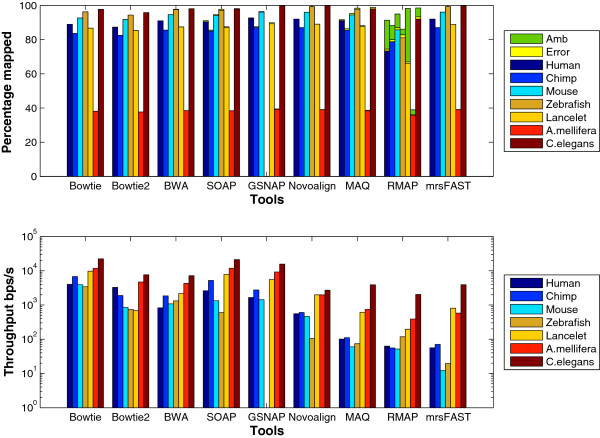
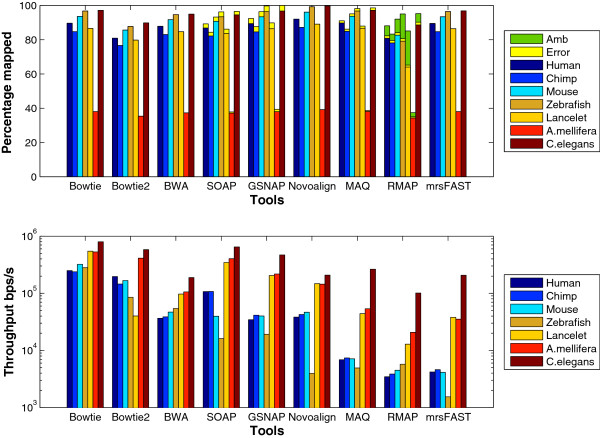
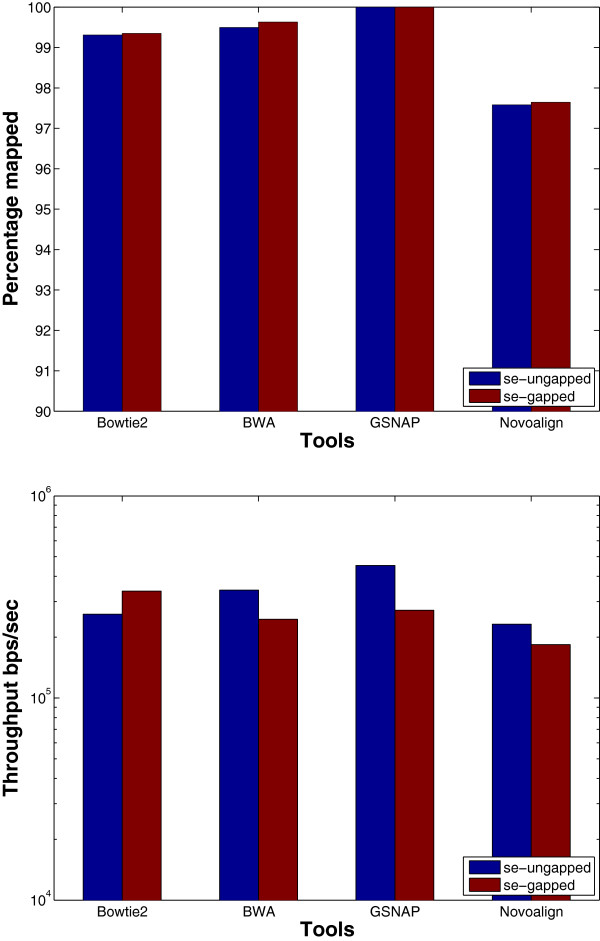
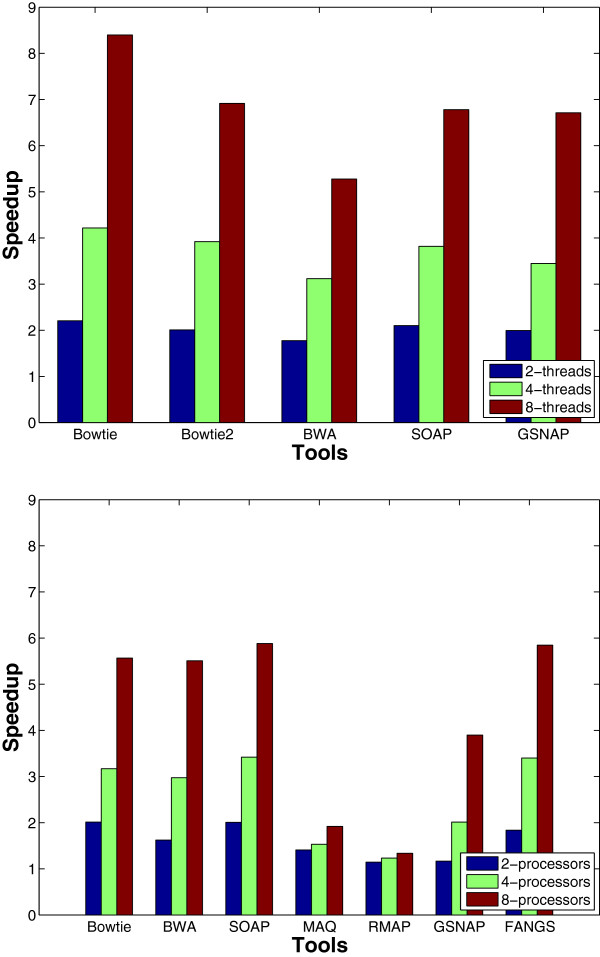
Results: We applied our benchmarking tests on 9 well known mapping tools, namely, Bowtie, Bowtie2, BWA, SOAP2, MAQ, RMAP, GSNAP, Novoalign, and mrsFAST (mrFAST) using synthetic data and real RNA-Seq data. MAQ and RMAP are based on building hash tables for the reads, whereas the remaining tools are based on indexing the reference genome. The benchmarking tests reveal the strengths and weaknesses of each tool. The results show that no single tool outperforms all others in all metrics. However, Bowtie maintained the best throughput for most of the tests while BWA performed better for longer read lengths. The benchmarking tests are not restricted to the mentioned tools and can be further applied to others.
Conclusion: The mapping process is still a hard problem that is affected by many factors. In this work, we provided a benchmarking suite that reveals and evaluates the different factors affecting the mapping process. Still, there is no tool that outperforms all of the others in all the tests. Therefore, the end user should clearly specify his needs in order to choose the tool that provides the best results.
Figures
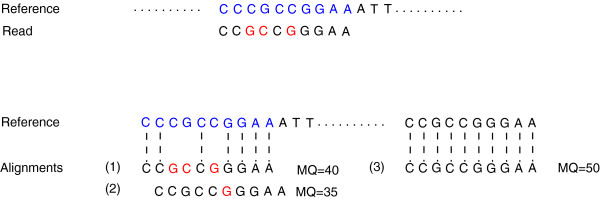
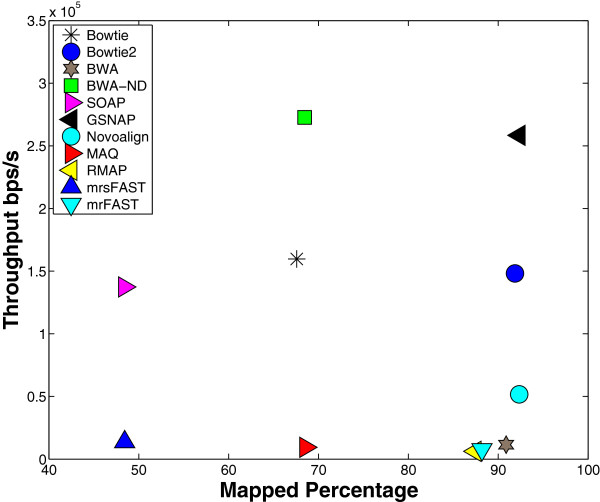
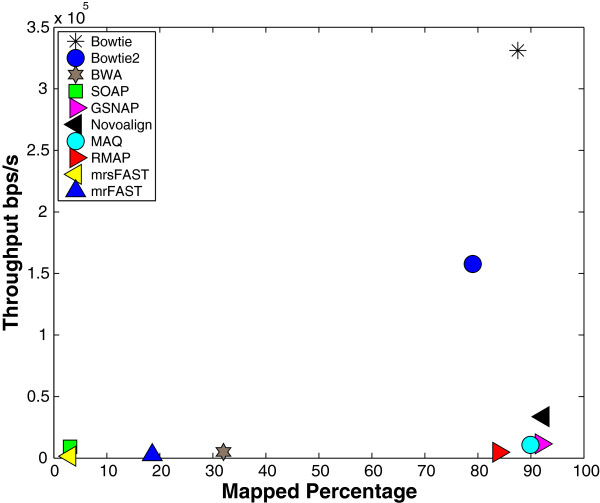
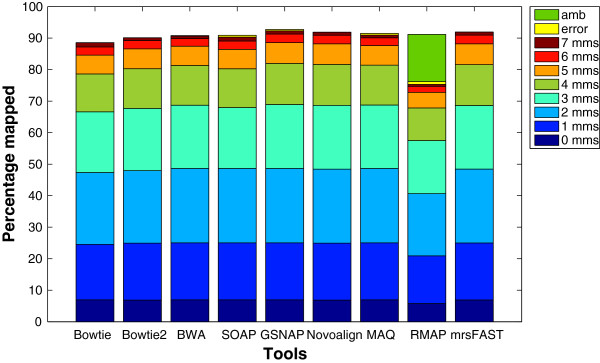
References
-
- National human genome institute. [ http://www.genome.gov]
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
