

A meta-analysis of the genomic and transcriptomic composition of complex life
- PMID: 23759593
- PMCID: PMC3737309
- DOI: 10.4161/cc.25134
A meta-analysis of the genomic and transcriptomic composition of complex life
Abstract
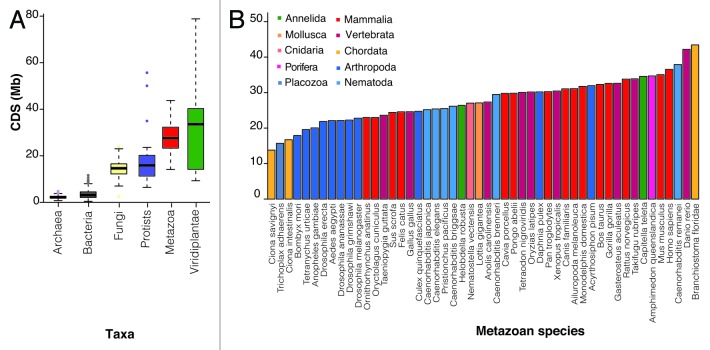
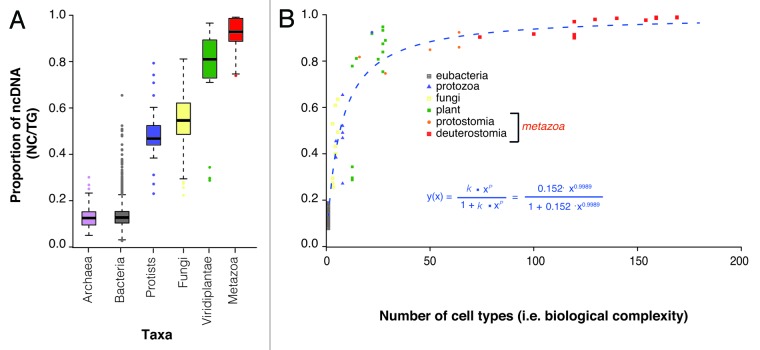
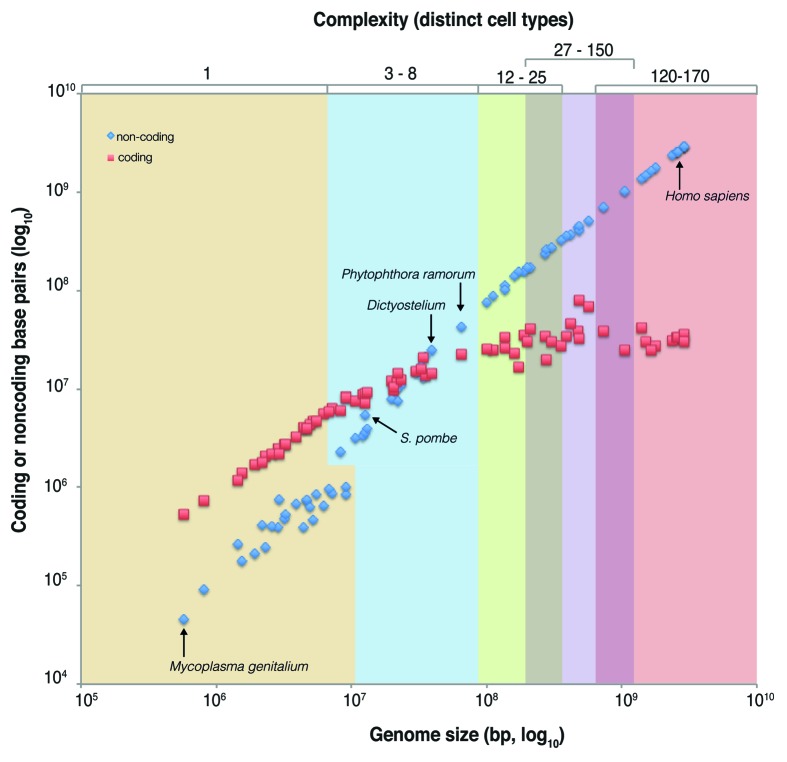
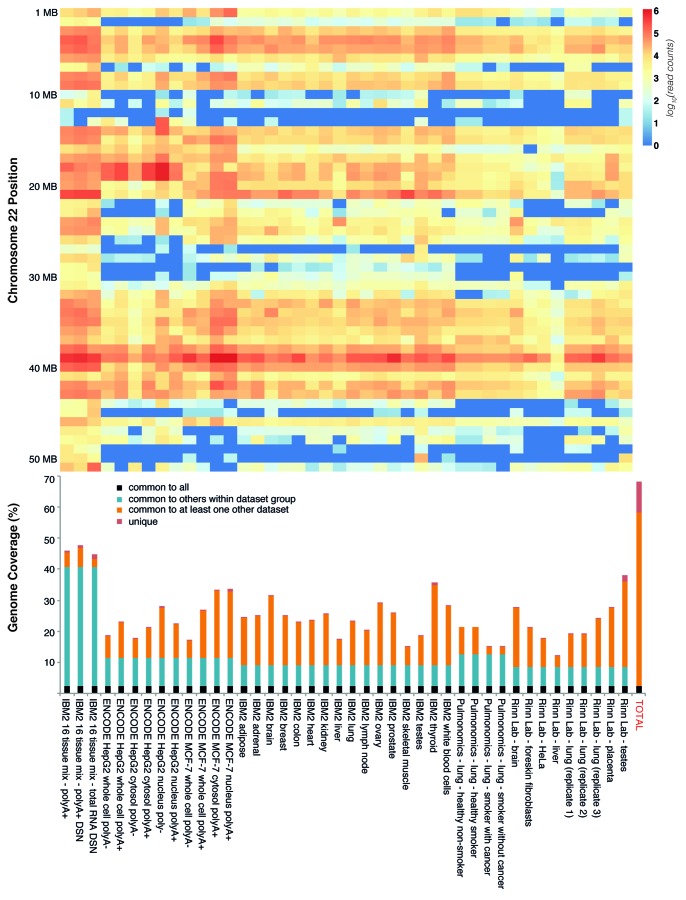
It is now clear that animal genomes are predominantly non-protein-coding, and that these sequences encode a wide array of RNA transcripts and other regulatory elements that are fundamental to the development of complex life. We have previously argued that the proportion of an animal genome that is non-protein-coding DNA (ncDNA) correlates well with its apparent biological complexity. Here we extend on that work and, using data from a total of 1,627 prokaryotic and 153 eukaryotic complete and annotated genomes, show that the proportion of ncDNA per haploid genome is significantly positively correlated with a previously published proxy of biological complexity, the number of distinct cell types. This is in contrast to the amount of the genome that encodes proteins, which we show is essentially unchanged across Metazoa. Furthermore, using a total of 179 RNA-seq data sets from nematode (47), fruit fly (72), zebrafish (20) and human (42), we show, consistent with other recent reports, that the vast majority of ncDNA in animals is transcribed. This includes more than 60 human loci previously considered "gene deserts," many of which are expressed tissue-specifically and associated with previously reported GWAS SNPs. These results suggest that ncDNA, and the ncRNAs encoded within it, may be intimately involved in the evolution, maintenance and development of complex life.
Keywords: complexity; evolution; lincRNA; non-coding DNA; noncoding RNA; small RNA.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources