

Cancer as a dysregulated epigenome allowing cellular growth advantage at the expense of the host
- PMID: 23760024
- PMCID: PMC4636434
- DOI: 10.1038/nrc3486
Cancer as a dysregulated epigenome allowing cellular growth advantage at the expense of the host
Abstract
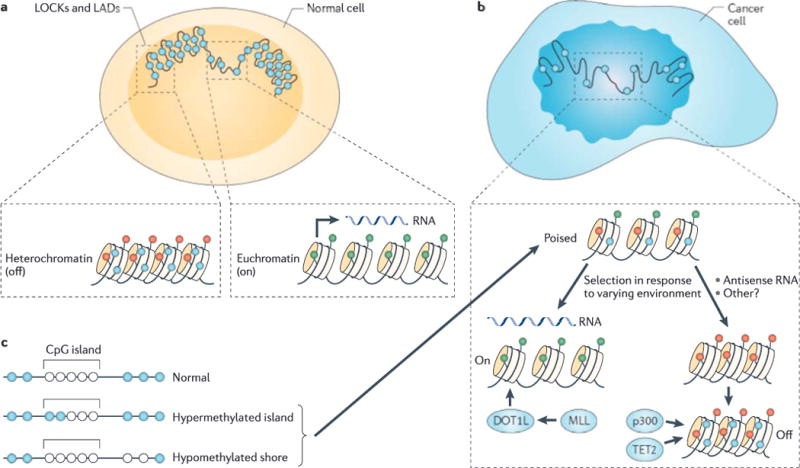
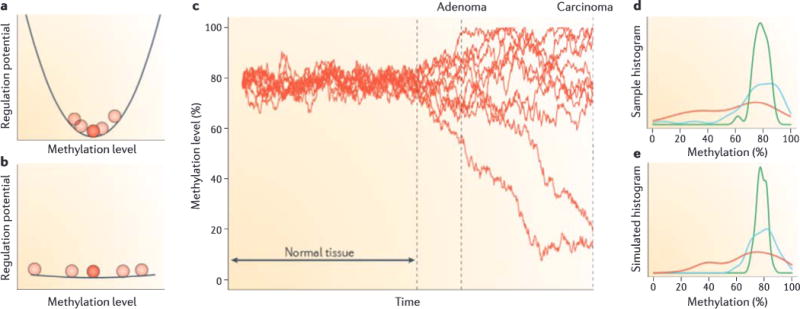
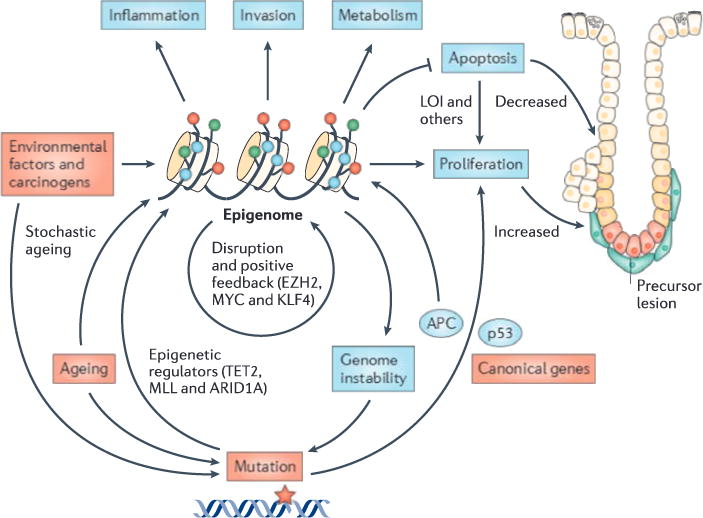
Although at the genetic level cancer is caused by diverse mutations, epigenetic modifications are characteristic of all cancers, from apparently normal precursor tissue to advanced metastatic disease, and these epigenetic modifications drive tumour cell heterogeneity. We propose a unifying model of cancer in which epigenetic dysregulation allows rapid selection for tumour cell survival at the expense of the host. Mechanisms involve both genetic mutations and epigenetic modifications that disrupt the function of genes that regulate the epigenome itself. Several exciting recent discoveries also point to a genome-scale disruption of the epigenome that involves large blocks of DNA hypomethylation, mutations of epigenetic modifier genes and alterations of heterochromatin in cancer (including large organized chromatin lysine modifications (LOCKs) and lamin-associated domains (LADs)), all of which increase epigenetic and gene expression plasticity. Our model suggests a new approach to cancer diagnosis and therapy that focuses on epigenetic dysregulation and has great potential for risk detection and chemoprevention.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
References
-
- Weinhouse S. Isozymes in cancer. Cancer Res. 1971;31:1166–1167. - PubMed
-
- Shih C, Weinberg RA. Isolation of a transforming sequence from a human bladder carcinoma cell line. Cell. 1982;29:161–169. - PubMed
-
- Feinberg AP, Vogelstein B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature. 1983;301:89–92. - PubMed
-
- Greger V, Passarge E, Hopping W, Messmer E, Horsthemke B. Epigenetic changes may contribute to the formation and spontaneous regression of retinoblastoma. Hum Genet. 1989;83:155–158. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
