

Review
doi: 10.1038/nrc3537.
Epub 2013 Jun 13.
End-joining, translocations and cancer
Affiliations
- PMID: 23760025
- PMCID: PMC5724777
- DOI: 10.1038/nrc3537
Item in Clipboard
Review
End-joining, translocations and cancer
Nat Rev Cancer.
2013 Jul.
Abstract
Fusion genes that are caused by chromosome translocations have been recognized for several decades as drivers of deregulated cell growth in certain types of cancer. In recent years, oncogenic fusion genes have been found in many haematological and solid tumours, demonstrating that translocations are a common cause of malignancy. Sequencing approaches have now confirmed that numerous, non-clonal translocations are a typical feature of cancer cells. These chromosome rearrangements are often highly complex and contain DNA sequence from multiple genomic sites. The factors and pathways that promote translocations are becoming clearer, with non-homologous end-joining implicated as a key source of genomic rearrangements.
Figures
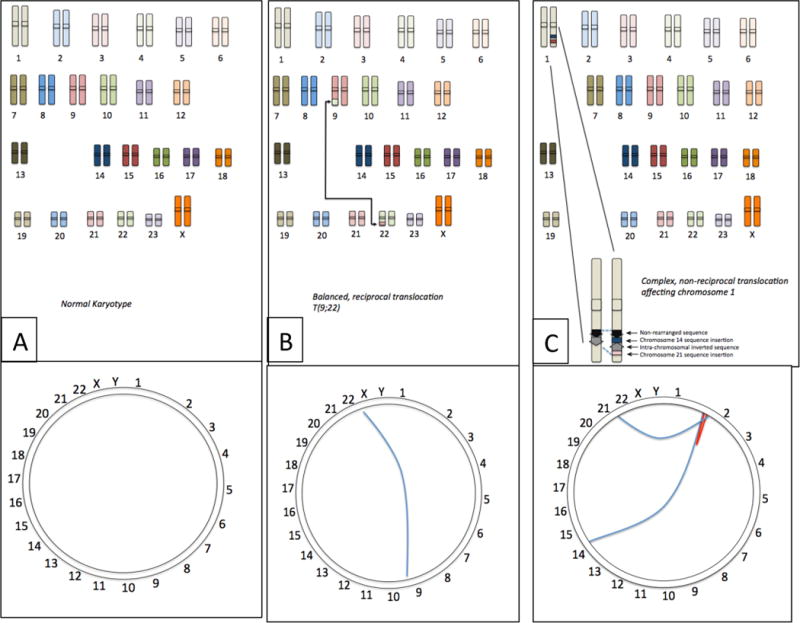
(A) The normal human chromosome set contains no rearrangements between chromosomes. The Circos plot shows this as a ring with the uninterrupted sequence of the chromosome running around the circumference. (B) Certain cancer cells contain balanced, reciprocal translocations, which join sequence from different chromosomes, such as the T(9;22) translocation from CLL, which exchanges sequence from chromosomes 9 and 22. Viewed as a Circos plot, this translocation can be visualized as a line connecting the breakpoints of the translocation on chromosomes 9 and 22. (C) Many translocations are more complex rearrangements involving multiple chromosomes. In this example, chromosome 1 contains a rearrangement involving translocated sequence from chromosomes 14 and 21, and an internal sequence inversion. Such complex translocations can be pictured using the Circos plot, where the blue lines indicate interchromosomal translocations and the red line shows the intra-chromosomal inversion.
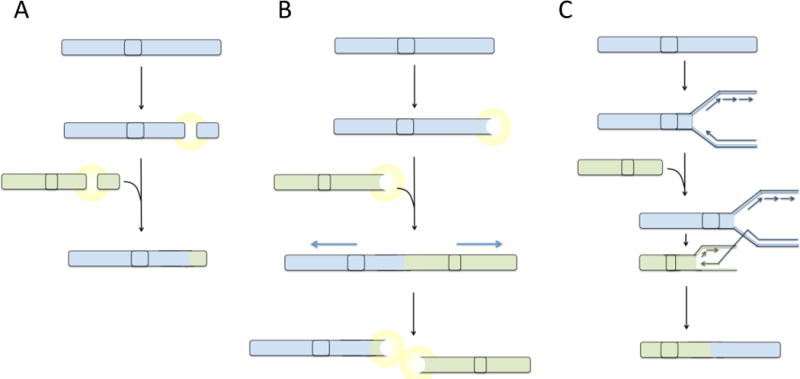
(A) Balanced reciprocal translocations are hypothesized to form as a consequence of fusion of two double-strand breaks that arise in the same cell. Following appearance of double-strand breaks, a signaling pathway is activated, which leads to ligation of the free DNA ends mediated by factors of the non-homologous end-joining pathway. (B) Telomere uncapping or attrition generates a DNA double-strand break response, potentially leading to fusion of telomeres generating end-to-end fusions. During anaphase, dicentric fusion chromosomes are pulled apart leading to the formation of translocations and double-strand breaks. Broken chromosomes act as substrates for additional rounds of fusion and breakage, generating increasingly complex translocations. (C) Hypothetically, translocations could arise by a replication-based mechanism by ‘switching’ of the DNA replication machinery to a site on a different chromosome with some degree of sequence homology to the original template. Extension of the replication fork at a site on a different chromosome would lead to a composite daughter strand being produced, containing sequence from two chromosomes. This composite chromosome would appear as a translocation. Highly complex translocations could be generated by multiple template switching events, generating an aberrant chromosome containing sequence from several different parts of the genome.
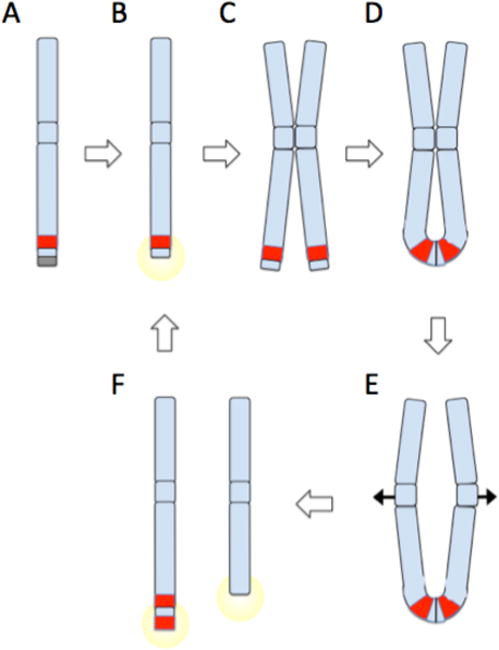
(A) Chromosomes are normally protected by telomeres (gray box). A sub-telomeric oncogene, shown in red, can become amplified by breakage fusion bridge cycles. (B) Telomere loss or double-strand breakage creates an unprotected DNA end, which triggers a DNA damage response. (C) Cancer cells with checkpoint defects will continue to grow despite DNA damage signaling, leading to duplication of the broken chromosome. (D) Ligation of broken chromatid ends produces an ‘anaphase bridge’, with a chromatin connection between the two sister chromatids. (E) As chromatids are drawn apart during anaphase, the anaphase bridge is subjected to increasing stress as centromeres are pulled to opposite poles of the dividing nucleus. Eventually, the anaphase bridge will shear, producing uneven derivative chromosomes as shown in (F). One derivative chromosome may capture sequence including a second copy of the oncogene from the broken sister chromatid. The broken chromosomes can act as substrates for further breakage fusion bridge cycles (B–F), potentially leading to dramatic amplification of oncogenes near telomeric sites. Oncogene amplification is a driver of malignant cell growth. If breakage fusion bridge cycles are combined with fusion of double strand breaks from other chromosomes, complex translocations can be built up featuring sequence from multiple chromosomes.
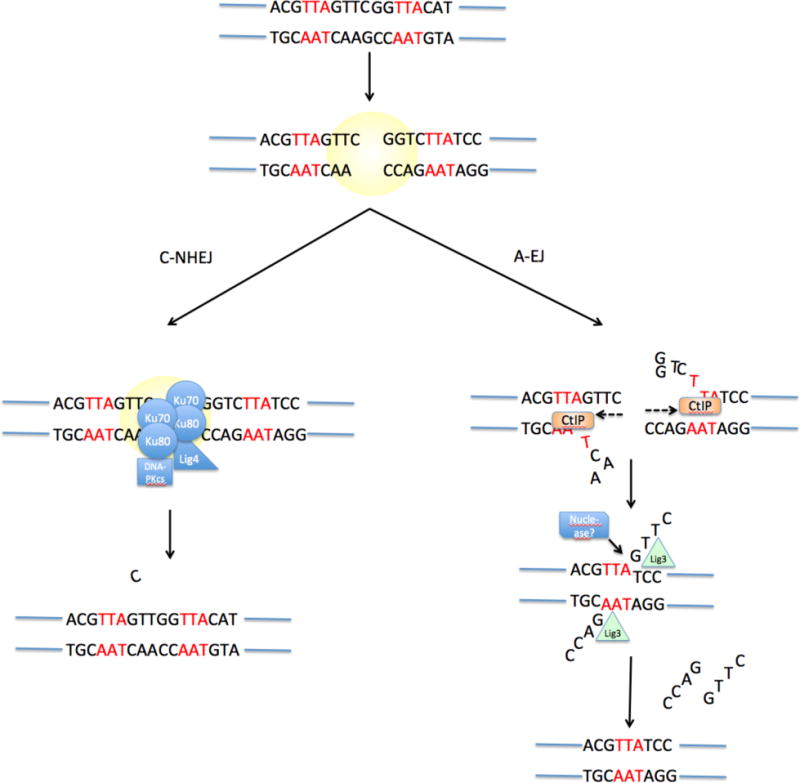
Upon appearance of a DNA double-strand break, two pathways can be active. Classical Non-Homologous End-Joining (C-NHEJ) involves binding of Ku70−Ku80 to the DNA break, followed by recruitment of DNA-PKcs and several other factors that mediate blunt-end ligation of the break by ligase 4 (LIG4). This process has no sequence requirements and may cause small-scale mutation such as the addition or deletion of a small number of nucleotides at the break junction. Alternative End-Joining (‘A-EJ’) involves exonucleolytic processing of the double-strand break to reveal stretches of potentially complementary sequence (microhomology, indicated in red) on either side of the break. This resection process may be mediated by the exonuclease CtIP. Following base-pairing at regions of microhomology, the ends are joined by an undetermined ligase enzyme (LIG).
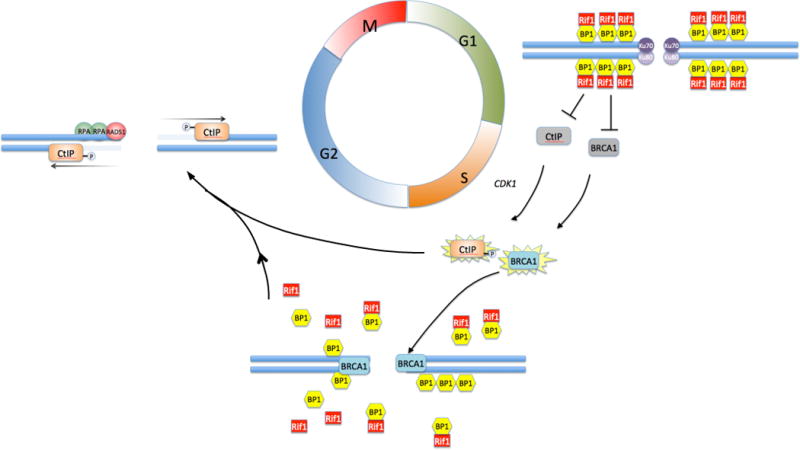
Non-homologous End-Joining is favored in G1, when the activities of BRCA1 and CtIP are repressed by a complex of 53BP1 and Rif1 that coats the chromatin in the vicinity of double-strand breaks. During the transition to S/G2, BRCA1 acquires the ability to bind at break sites despite the repressive effect of 53BP1 and Rif1. The mechanism for BRCA1 activation and recruitment is still unknown. Depletion of Rif1 and activation by cyclin dependent kinase 1 (CDK1)-mediated phosphorylation allows CtIP to become active at the break site, where it resects duplex DNA to form a 5′ single-strand overhang. This favors resection-dependent repair pathways, including A-EJ and HR. Commitment to HR is mediated by loading of Replication Protein A (RPA) and Rad51 at the break site.
References
-
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. - PubMed
-
- Forment JV, Kaidi A, Jackson SP. Chromothripsis and cancer: causes and consequences of chromosome shattering. Nat Rev Cancer. 2012;12:663–70. - PubMed
-
- Giardino D, et al. De novo balanced chromosome rearrangements in prenatal diagnosis. Prenat Diagn. 2009;29:257–65. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
