

Low density lipoprotein receptor-related protein 1 (LRP1) modulates N-methyl-D-aspartate (NMDA) receptor-dependent intracellular signaling and NMDA-induced regulation of postsynaptic protein complexes
- PMID: 23760271
- PMCID: PMC3724646
- DOI: 10.1074/jbc.M112.444364
Low density lipoprotein receptor-related protein 1 (LRP1) modulates N-methyl-D-aspartate (NMDA) receptor-dependent intracellular signaling and NMDA-induced regulation of postsynaptic protein complexes
Abstract
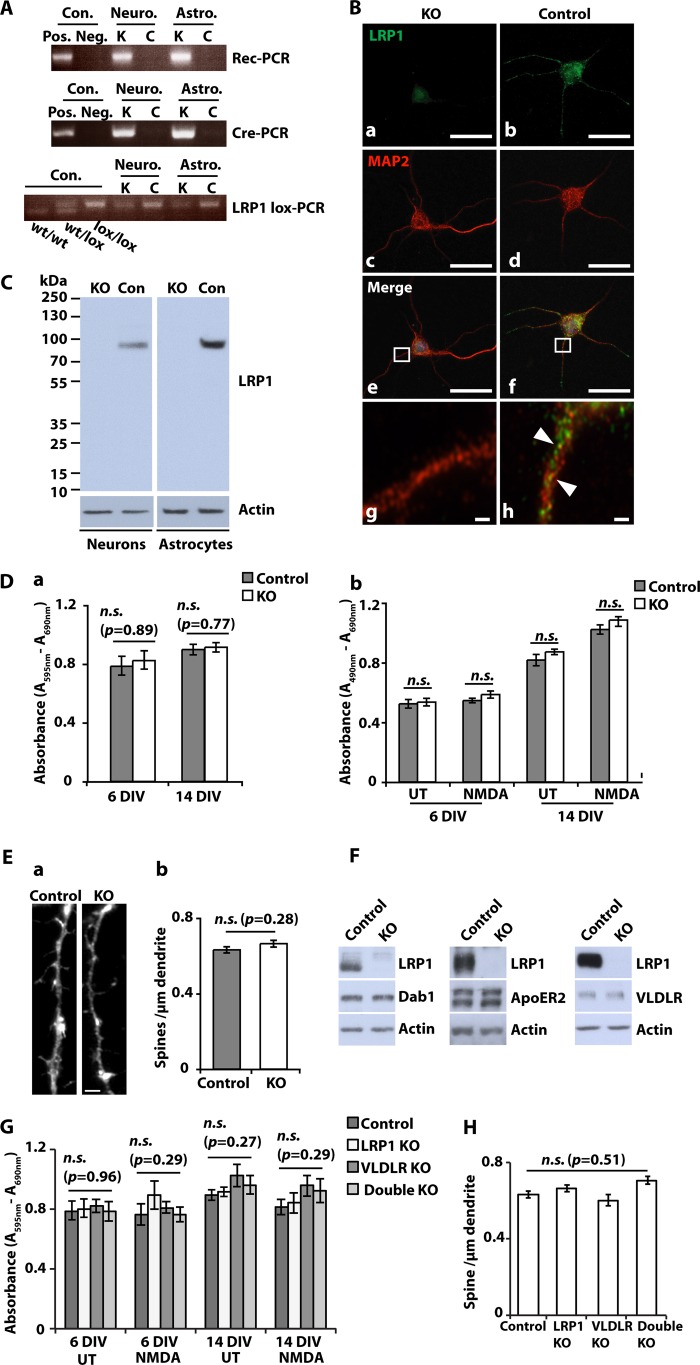
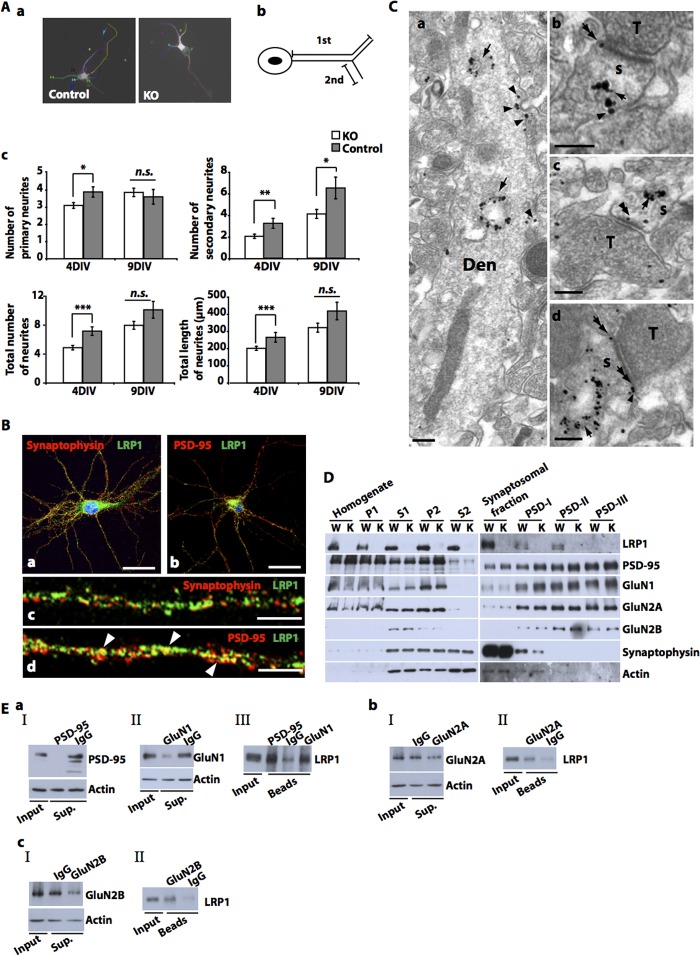
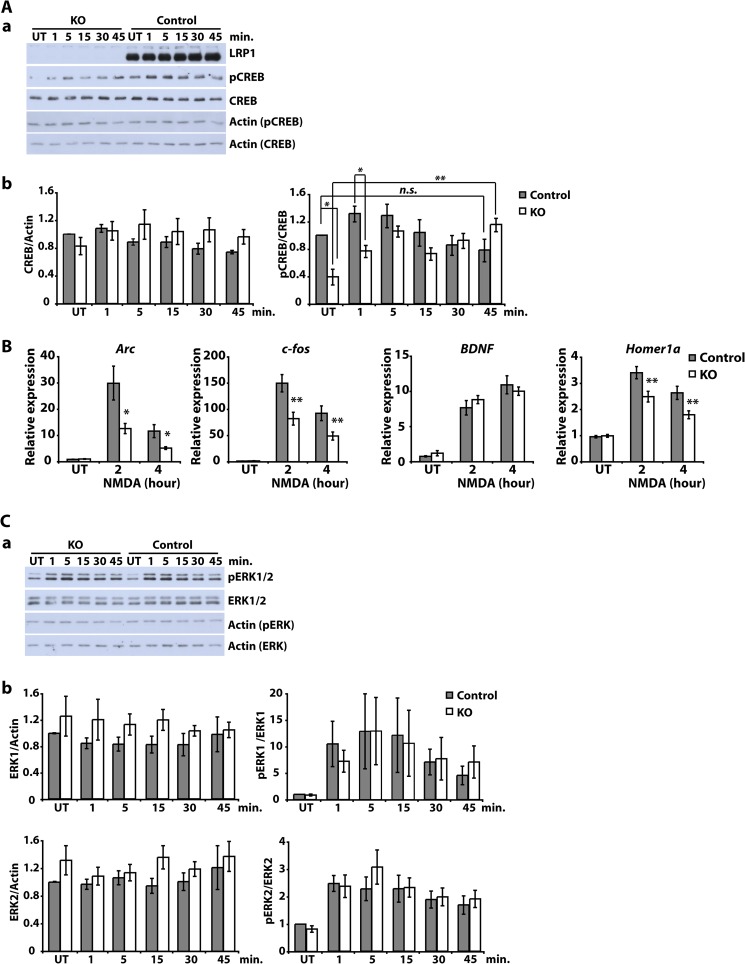
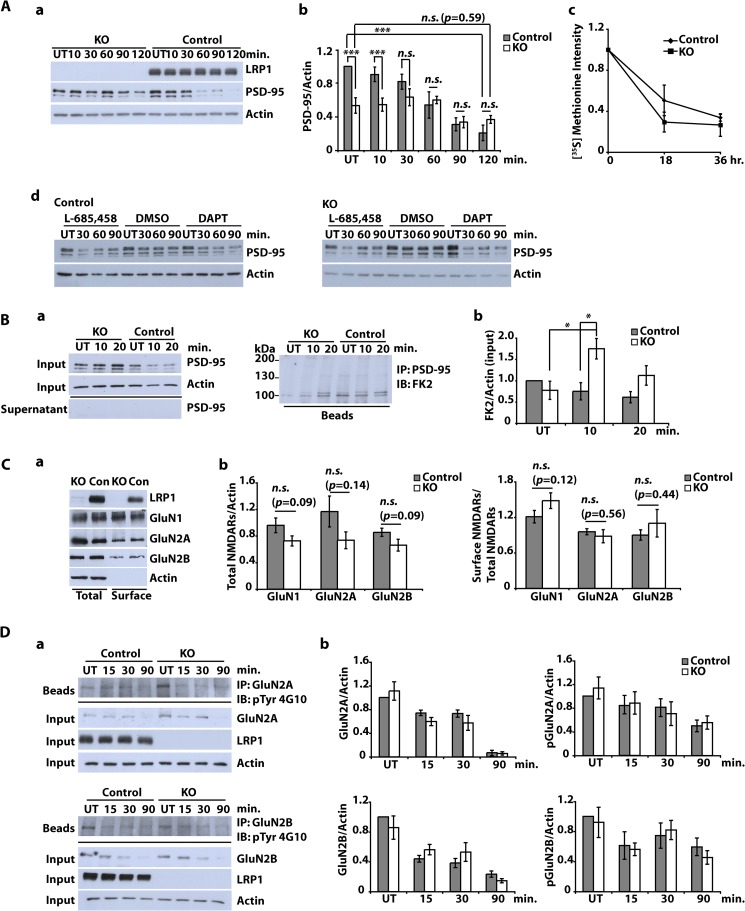
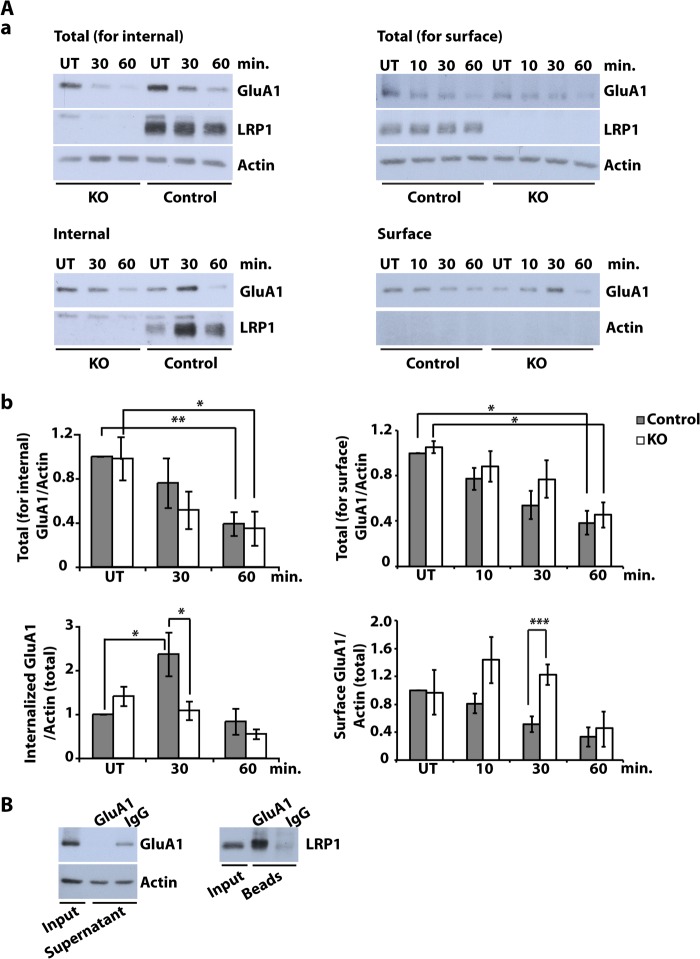
The lipoprotein receptor LRP1 is essential in neurons of the central nervous system, as was revealed by the analysis of conditional Lrp1-deficient mouse models. The molecular basis of its neuronal functions, however, is still incompletely understood. Here we show by immunocytochemistry, electron microscopy, and postsynaptic density preparation that LRP1 is located postsynaptically. Basal and NMDA-induced phosphorylation of the transcription factor cAMP-response element-binding protein (CREB) as well as NMDA target gene transcription are reduced in LRP1-deficient neurons. In control neurons, NMDA promotes γ-secretase-dependent release of the LRP1 intracellular domain (LRP1-ICD). However, pull-down and chromatin immunoprecipitation (ChIP) assays showed no direct interaction between the LRP1-ICD and either CREB or target gene promoters. On the other hand, NMDA-induced degradation of the postsynaptic scaffold protein PSD-95 was impaired in the absence of LRP1, whereas its ubiquitination was increased, indicating that LRP1 influences the composition of postsynaptic protein complexes. Accordingly, NMDA-induced internalization of the AMPA receptor subunit GluA1 was impaired in LRP1-deficient neurons. These results show a role of LRP1 in the regulation and turnover of synaptic proteins, which may contribute to the reduced dendritic branching and to the neurological phenotype observed in the absence of LRP1.
Keywords: Glutamate Receptors; Lipoprotein Receptor; Secretases; Signaling; Synapses.
Figures

References
-
- May P., Rohlmann A., Bock H. H., Zurhove K., Marth J. D., Schomburg E. D., Noebels J. L., Beffert U., Sweatt J. D., Weeber E. J., Herz J. (2004) Neuronal LRP1 functionally associates with postsynaptic proteins and is required for normal motor function in mice. Mol. Cell. Biol. 24, 8872–8883 - PMC - PubMed
-
- Kang D. E., Pietrzik C. U., Baum L., Chevallier N., Merriam D. E., Kounnas M. Z., Wagner S. L., Troncoso J. C., Kawas C. H., Katzman R., Koo E. H. (2000) Modulation of amyloid β-protein clearance and Alzheimer's disease susceptibility by the LDL receptor-related protein pathway. J. Clin. Invest. 106, 1159–1166 - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
