

Increased in vivo amyloid-β42 production, exchange, and loss in presenilin mutation carriers
- PMID: 23761040
- PMCID: PMC3838868
- DOI: 10.1126/scitranslmed.3005615
Increased in vivo amyloid-β42 production, exchange, and loss in presenilin mutation carriers
Abstract
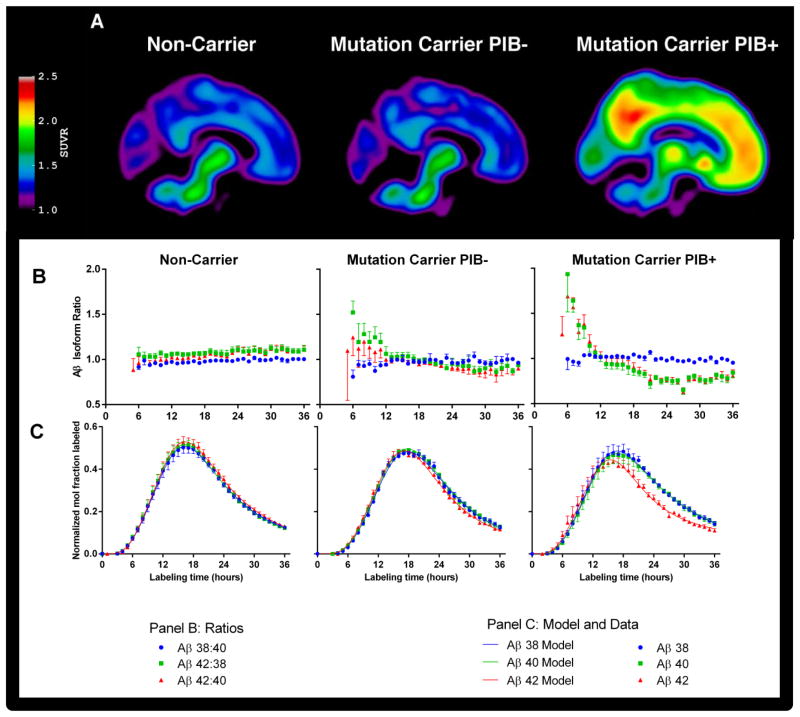
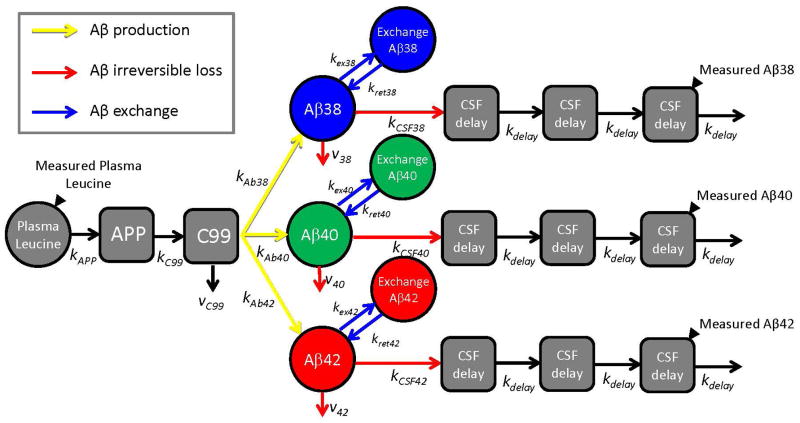
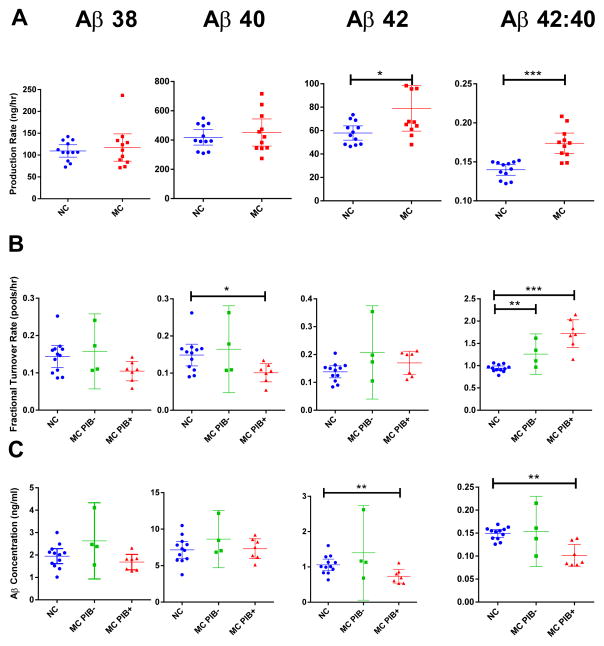
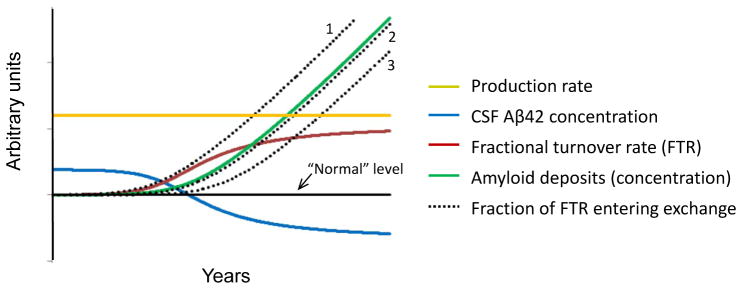
Alzheimer's disease (AD) is hypothesized to be caused by an overproduction or reduced clearance of amyloid-β (Aβ) peptide. Autosomal dominant AD (ADAD) caused by mutations in the presenilin (PSEN) gene have been postulated to result from increased production of Aβ42 compared to Aβ40 in the central nervous system (CNS). This has been demonstrated in rodent models of ADAD but not in human mutation carriers. We used compartmental modeling of stable isotope labeling kinetic (SILK) studies in human carriers of PSEN mutations and related noncarriers to evaluate the pathophysiological effects of PSEN1 and PSEN2 mutations on the production and turnover of Aβ isoforms. We compared these findings by mutation status and amount of fibrillar amyloid deposition as measured by positron emission tomography (PET) using the amyloid tracer Pittsburgh compound B (PIB). CNS Aβ42 to Aβ40 production rates were 24% higher in mutation carriers compared to noncarriers, and this was independent of fibrillar amyloid deposits quantified by PET PIB imaging. The fractional turnover rate of soluble Aβ42 relative to Aβ40 was 65% faster in mutation carriers and correlated with amyloid deposition, consistent with increased deposition of Aβ42 into plaques, leading to reduced recovery of Aβ42 in cerebrospinal fluid (CSF). Reversible exchange of Aβ42 peptides with preexisting unlabeled peptide was observed in the presence of plaques. These findings support the hypothesis that Aβ42 is overproduced in the CNS of humans with PSEN mutations that cause AD, and demonstrate that soluble Aβ42 turnover and exchange processes are altered in the presence of amyloid plaques, causing a reduction in Aβ42 concentrations in the CSF.
Conflict of interest statement
TB has served on an advisory board for Eli Lilly and has received research funding from Avid Radiopharmaceuticals. These relationships are not related to the content in the manuscript.
AG has received research funding during the last 12 months from Pfizer, Genentech, AstraZeneca and iPierian and has served as a consultant for Amgen. These relationships are not related to the content in the manuscript.
JM serves on scientific advisory boards for Eisai, Esteve, Janssen Alzheimer Immunotherapy Program, Glaxo-Smith-Kline, Novartis, and Pfizer.
The other authors declare that they have no competing interests.
Figures
References
-
- Bateman RJ, Xiong C, Benzinger TLS, Fagan AM, Goate A, Fox NC, Marcus DS, Cairns NJ, Xie X, Blazey TM, Holtzman DM, Santacruz A, Buckles V, Oliver A, Moulder K, Aisen PS, Ghetti B, Klunk WE, McDade E, Martins RN, Masters CL, Mayeux R, Ringman JM, Rossor MN, Schofield PR, Sperling RA, Salloway S, Morris JC. Clinical and Biomarker Changes in Dominantly Inherited Alzheimer’s Disease. N Engl J Med. 2012:367. - PMC - PubMed
-
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002:297. - PubMed
-
- Haass C, De Strooper B. The presenilins in Alzheimer’s disease--proteolysis holds the key. Science. 1999:286. - PubMed
-
- Wang R, Sweeney D, Gandy SE, Sisodia SS. The profile of soluble amyloid beta protein in cultured cell media. Detection and quantification of amyloid beta protein and variants by immunoprecipitation-mass spectrometry. J Biol Chem. 1996:271. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- U24 AG021886/AG/NIA NIH HHS/United States
- P41 GM103422/GM/NIGMS NIH HHS/United States
- 5P01AG026276-S1/AG/NIA NIH HHS/United States
- UL1 TR000448/TR/NCATS NIH HHS/United States
- P60 DK020579/DK/NIDDK NIH HHS/United States
- P30 DK020579/DK/NIDDK NIH HHS/United States
- P41 RR000954/RR/NCRR NIH HHS/United States
- R24 GM136766/GM/NIGMS NIH HHS/United States
- R01 NS065667/NS/NINDS NIH HHS/United States
- R01-NS065667/NS/NINDS NIH HHS/United States
- P30 DK056341/DK/NIDDK NIH HHS/United States
- P01 AG026276/AG/NIA NIH HHS/United States
- 5P01 AG026272/AG/NIA NIH HHS/United States
- UL1 RR024992/RR/NCRR NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
