

Novel mutations in the SCNN1A gene causing Pseudohypoaldosteronism type 1
- PMID: 23762408
- PMCID: PMC3675083
- DOI: 10.1371/journal.pone.0065676
Novel mutations in the SCNN1A gene causing Pseudohypoaldosteronism type 1
Abstract
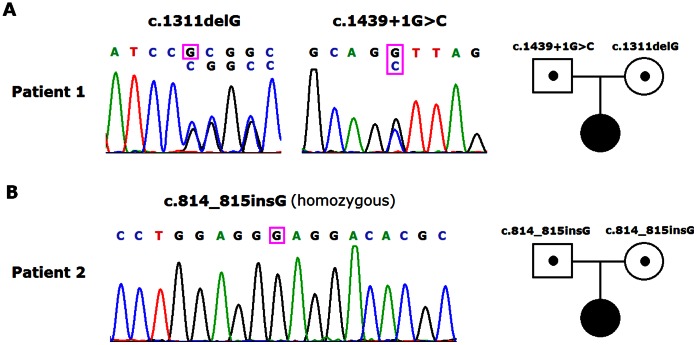
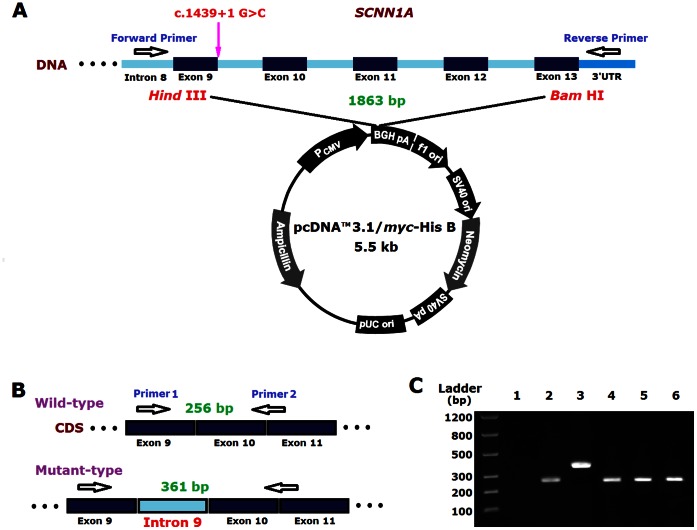
Pseudohypoaldosteronism type 1 (PHA1) is a rare inherited disease characterized by resistance to the actions of aldosterone. Mutations in the subunit genes (SCNN1A, SCNN1B, SCNN1G) of the epithelial sodium channel (ENaC) and the NR3C2 gene encoding the mineralocorticoid receptor, result in systemic PHA1 and renal PHA1 respectively. Common clinical manifestations of PHA1 include salt wasting, hyperkalaemia, metabolic acidosis and elevated plasma aldosterone levels in the neonatal period. In this study, we describe the clinical and biochemical manifestations in two Chinese patients with systemic PHA1. Sequence analysis of the SCNN1A gene revealed a compound heterozygous mutation (c.1311delG and c.1439+1G>C) in one patient and a homozygous mutation (c.814_815insG) in another patient, all three variants are novel. Further analysis of the splicing pattern in a minigene construct showed that the c.1439+1G>C mutation can lead to the retainment of intron 9 as the 5'-donor splice site disappears during post-transcriptional processing of mRNA. In conclusion, our study identified three novel SCNN1A gene mutations in two Chinese patients with systemic PHA1.
Conflict of interest statement
Figures
References
-
- Riepe FG (2009) Clinical and molecular features of type 1 pseudohypoaldosteronism. Horm Res 72: 1–9. - PubMed
-
- Furgeson SB, Linas S (2010) Mechanisms of type I and type II pseudohypoaldosteronism. J Am Soc Nephrol 21: 1842–1845. - PubMed
-
- Dirlewanger M, Huser D, Zennaro MC, Girardin E, Schild L, et al. (2011) A homozygous missense mutation in SCNN1A is responsible for a transient neonatal form of pseudohypoaldosteronism type 1. Am J Physiol Endocrinol Metab 301: E467–73. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
