

A modified RNA-Seq approach for whole genome sequencing of RNA viruses from faecal and blood samples
- PMID: 23762474
- PMCID: PMC3677912
- DOI: 10.1371/journal.pone.0066129
A modified RNA-Seq approach for whole genome sequencing of RNA viruses from faecal and blood samples
Abstract
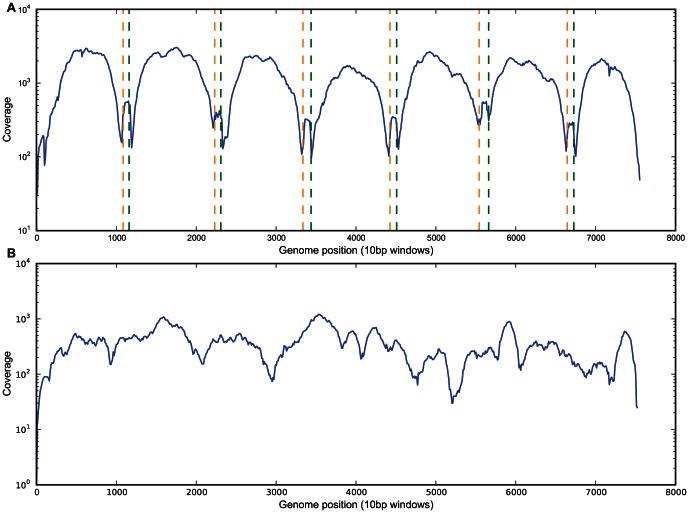
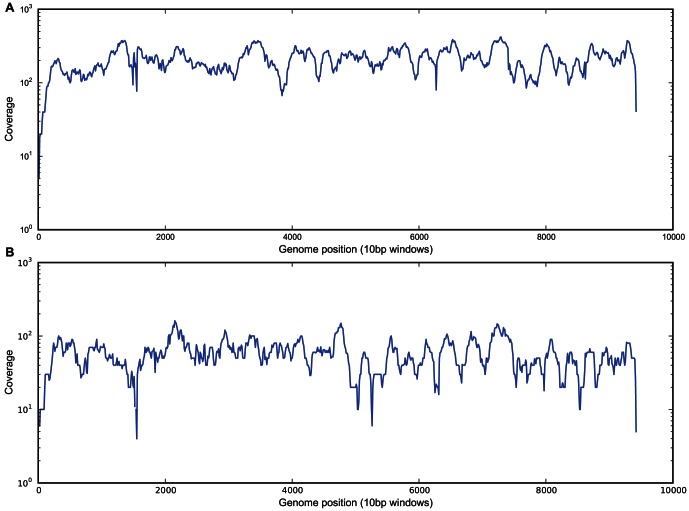
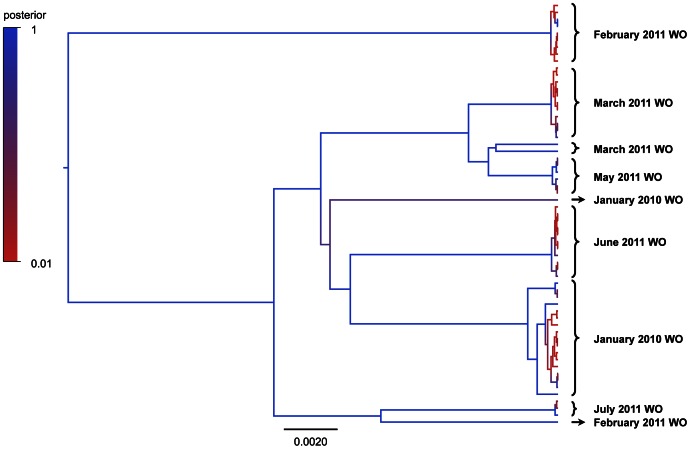
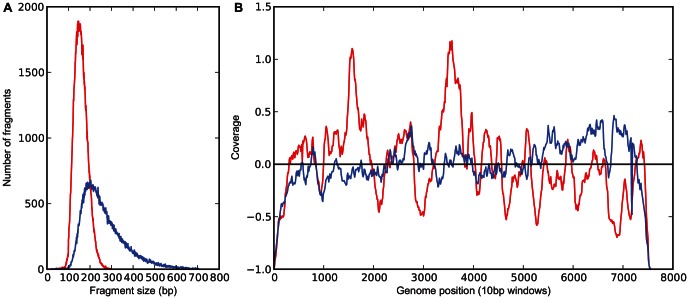
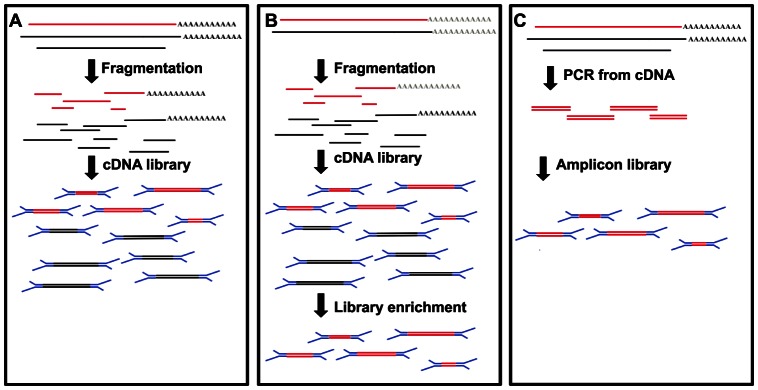
To date, very large scale sequencing of many clinically important RNA viruses has been complicated by their high population molecular variation, which creates challenges for polymerase chain reaction and sequencing primer design. Many RNA viruses are also difficult or currently not possible to culture, severely limiting the amount and purity of available starting material. Here, we describe a simple, novel, high-throughput approach to Norovirus and Hepatitis C virus whole genome sequence determination based on RNA shotgun sequencing (also known as RNA-Seq). We demonstrate the effectiveness of this method by sequencing three Norovirus samples from faeces and two Hepatitis C virus samples from blood, on an Illumina MiSeq benchtop sequencer. More than 97% of reference genomes were recovered. Compared with Sanger sequencing, our method had no nucleotide differences in 14,019 nucleotides (nt) for Noroviruses (from a total of 2 Norovirus genomes obtained with Sanger sequencing), and 8 variants in 9,542 nt for Hepatitis C virus (1 variant per 1,193 nt). The three Norovirus samples had 2, 3, and 2 distinct positions called as heterozygous, while the two Hepatitis C virus samples had 117 and 131 positions called as heterozygous. To confirm that our sample and library preparation could be scaled to true high-throughput, we prepared and sequenced an additional 77 Norovirus samples in a single batch on an Illumina HiSeq 2000 sequencer, recovering >90% of the reference genome in all but one sample. No discrepancies were observed across 118,757 nt compared between Sanger and our custom RNA-Seq method in 16 samples. By generating viral genomic sequences that are not biased by primer-specific amplification or enrichment, this method offers the prospect of large-scale, affordable studies of RNA viruses which could be adapted to routine diagnostic laboratory workflows in the near future, with the potential to directly characterize within-host viral diversity.
Conflict of interest statement
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
