

Computational analysis of four human adenovirus type 4 genomes reveals molecular evolution through two interspecies recombination events
- PMID: 23763770
- PMCID: PMC3779658
- DOI: 10.1016/j.virol.2013.05.014
Computational analysis of four human adenovirus type 4 genomes reveals molecular evolution through two interspecies recombination events
Abstract
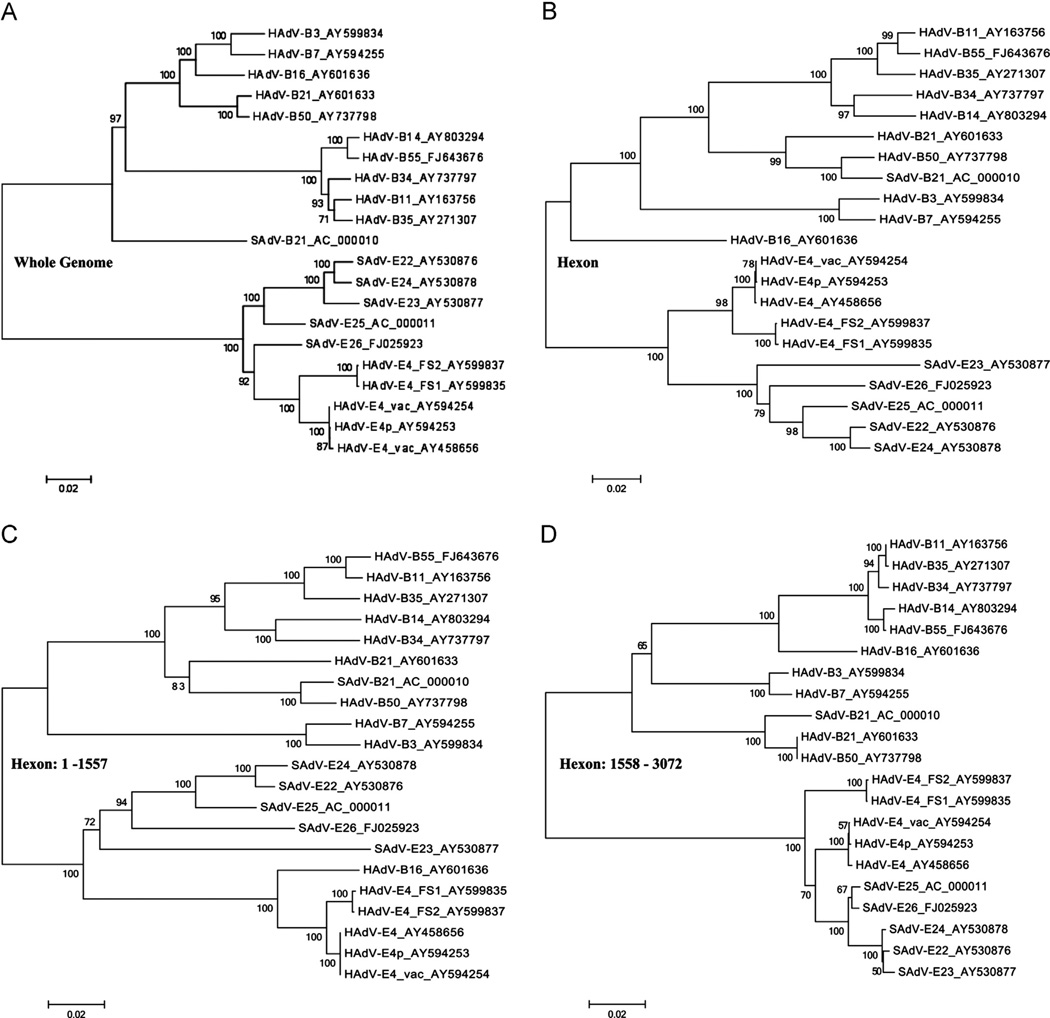
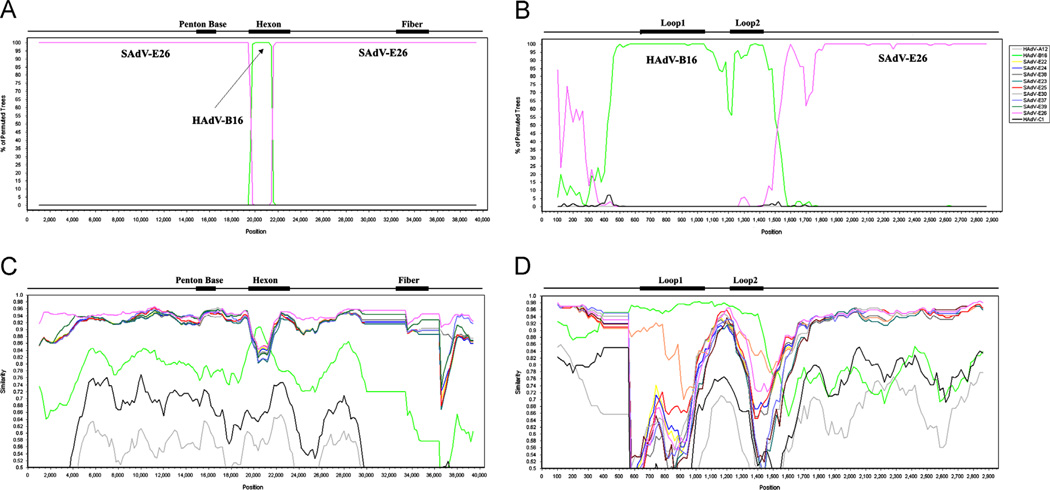
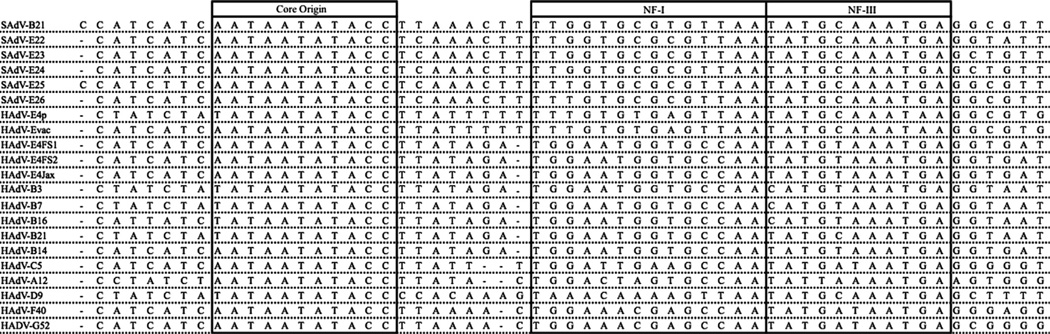
Computational analysis of human adenovirus type 4 (HAdV-E4), a pathogen that is the only HAdV member of species E, provides insights into its zoonotic origin and molecular adaptation. Its genome encodes a domain of the major capsid protein, hexon, from HAdV-B16 recombined into the genome chassis of a simian adenovirus. Genomes of two recent field strains provide a clue to its adaptation to the new host: recombination of a NF-I binding site motif, which is required for efficient viral replication, from another HAdV genome. This motif is absent in the chimpanzee adenoviruses and the HAdV-E4 prototype, but is conserved amongst other HAdVs. This is the first report of an interspecies recombination event for HAdVs, and the first documentation of a lateral partial gene transfer from a chimpanzee AdV. The potential for such recombination events are important when considering chimpanzee adenoviruses as candidate gene delivery vectors for human patients.
Keywords: Adenovirus; Molecular evolution; Recombination; Zoonosis.
Copyright © 2013 Elsevier Inc. All rights reserved.
Figures

References
-
- Adrian T, Wadell G, Hierholzer JC, Wigand R. DNA restriction analysis ol adenovirus prototypes 1 to 41. Arch. Virol. 1986;91:277–290. - PubMed
-
- Bailey A, Mautner V. Phylogenetic relationships among adenovirus serotypes. Virology. 1994;205:438–52. - PubMed
-
- Barrero PR, Valinotto LE, Tittarelli E, Mistchenko AS. Molecular typing of adenoviruses in pediatric respiratory infections in Buenos Aires, Argentina (1999–2010) J. Clin. Virol. 2012;53:145–150. - PubMed
-
- Basnight M, Jr., Rogers NG, Gibbs CJ, Jr., Gajdusek DC. Characterization of four new adenovirus serotypes isolated from chimpanzee tissue explants. Am. J. Epidemiol. 1971;94:166–171. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
