

Folding the proteome
- PMID: 23764454
- PMCID: PMC3691291
- DOI: 10.1016/j.tibs.2013.05.001
Folding the proteome
Abstract
Protein folding is an essential prerequisite for protein function and hence cell function. Kinetic and thermodynamic studies of small proteins that refold reversibly were essential for developing our current understanding of the fundamentals of protein folding mechanisms. However, we still lack sufficient understanding to accurately predict protein structures from sequences, or the effects of disease-causing mutations. To date, model proteins selected for folding studies represent only a small fraction of the complexity of the proteome and are unlikely to exhibit the breadth of folding mechanisms used in vivo. We are in urgent need of new methods - both theoretical and experimental - that can quantify the folding behavior of a truly broad set of proteins under in vivo conditions. Such a shift in focus will provide a more comprehensive framework from which to understand the connections between protein folding, the molecular basis of disease, and cell function and evolution.
Copyright © 2013 Elsevier Ltd. All rights reserved.
Figures
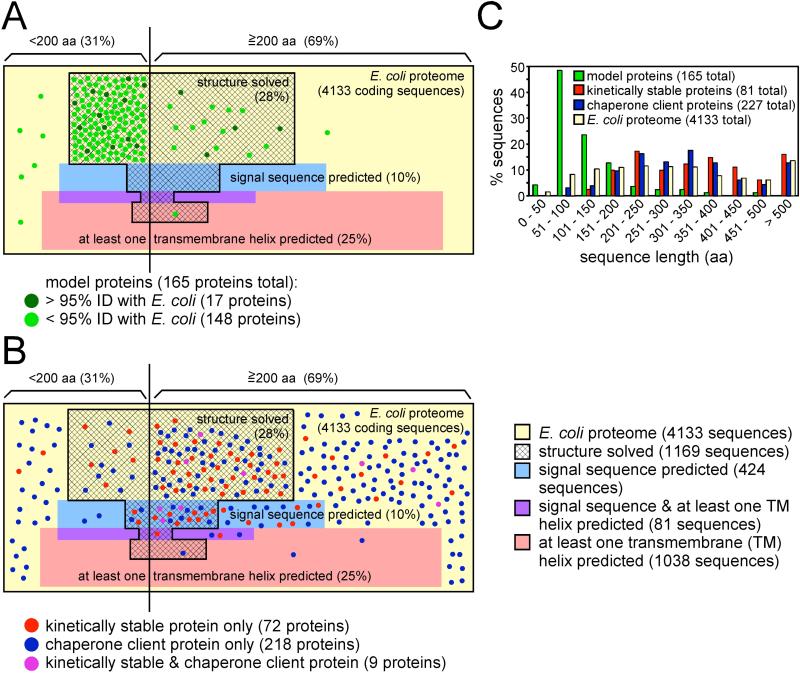
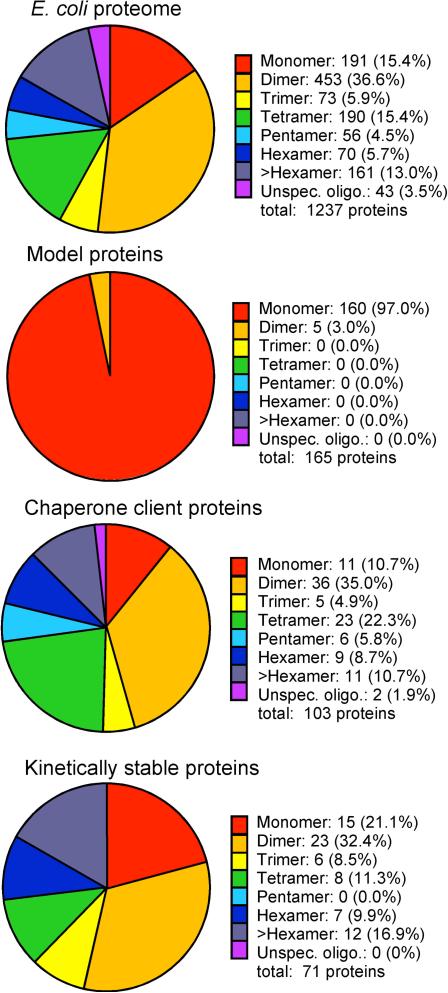
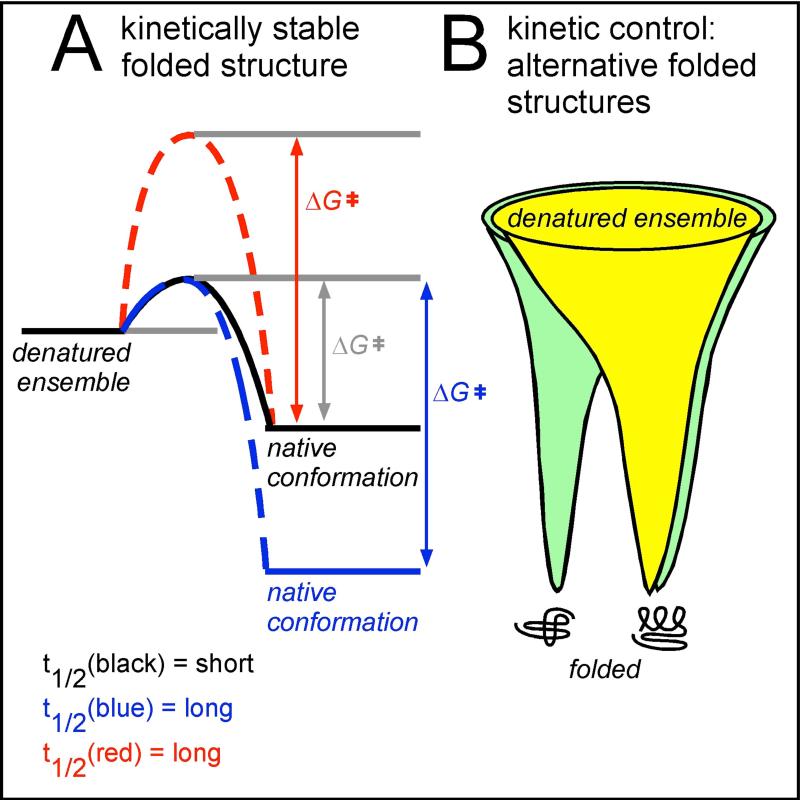
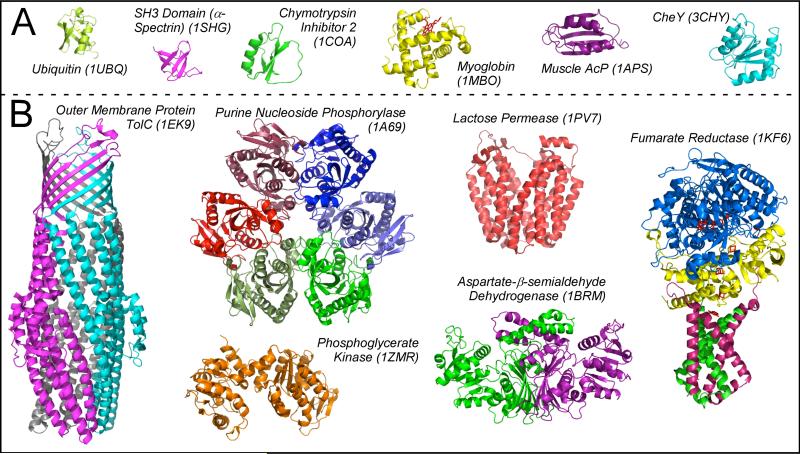
References
-
- Anfinsen CB. Principles that govern the folding of protein chains. Science. 1973;181:223–230. - PubMed
-
- Pace CN. Determination and analysis of urea and guanidine hydrochloride denaturation curves. Methods Enzymol. 1986;131:266–280. - PubMed
-
- Plaxco KW, et al. Contact order, transition state placement and the refolding rates of single domain proteins. J. Mol. Biol. 1998;277:985–994. - PubMed
-
- Jackson SE. How do small single-domain proteins fold? Fold. Des. 1998;3:R81–R91. - PubMed
-
- Galzitskaya OV, et al. Chain length is the main determinant of the folding rate for proteins with three-state folding kinetics. Proteins. 2003;51:162–166. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
