

General framework for meta-analysis of rare variants in sequencing association studies
- PMID: 23768515
- PMCID: PMC3710762
- DOI: 10.1016/j.ajhg.2013.05.010
General framework for meta-analysis of rare variants in sequencing association studies
Abstract
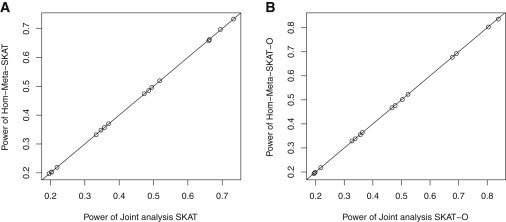
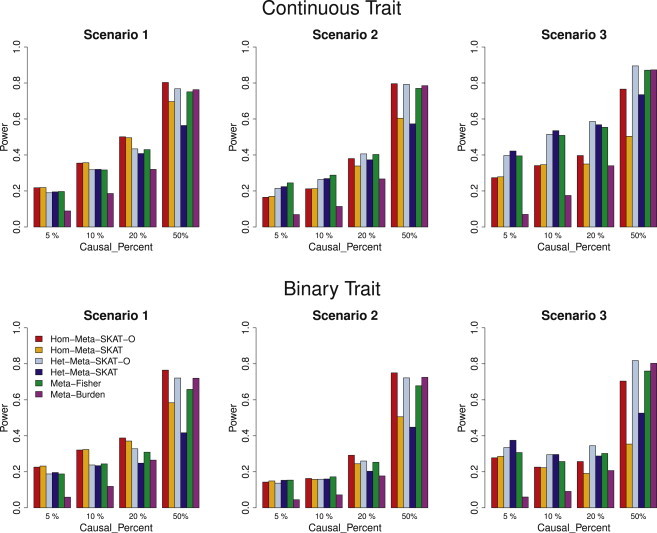
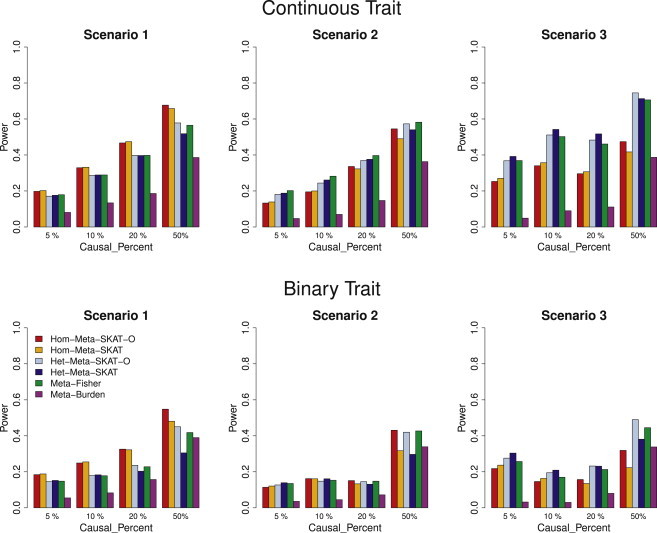
We propose a general statistical framework for meta-analysis of gene- or region-based multimarker rare variant association tests in sequencing association studies. In genome-wide association studies, single-marker meta-analysis has been widely used to increase statistical power by combining results via regression coefficients and standard errors from different studies. In analysis of rare variants in sequencing studies, region-based multimarker tests are often used to increase power. We propose meta-analysis methods for commonly used gene- or region-based rare variants tests, such as burden tests and variance component tests. Because estimation of regression coefficients of individual rare variants is often unstable or not feasible, the proposed method avoids this difficulty by calculating score statistics instead that only require fitting the null model for each study and then aggregating these score statistics across studies. Our proposed meta-analysis rare variant association tests are conducted based on study-specific summary statistics, specifically score statistics for each variant and between-variant covariance-type (linkage disequilibrium) relationship statistics for each gene or region. The proposed methods are able to incorporate different levels of heterogeneity of genetic effects across studies and are applicable to meta-analysis of multiple ancestry groups. We show that the proposed methods are essentially as powerful as joint analysis by directly pooling individual level genotype data. We conduct extensive simulations to evaluate the performance of our methods by varying levels of heterogeneity across studies, and we apply the proposed methods to meta-analysis of rare variant effects in a multicohort study of the genetics of blood lipid levels.
Copyright © 2013 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Ansorge W.J. Next-generation DNA sequencing techniques. New Biotechnol. 2009;25:195–203. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
