

Viral modulation of programmed necrosis
- PMID: 23773332
- PMCID: PMC3821070
- DOI: 10.1016/j.coviro.2013.05.019
Viral modulation of programmed necrosis
Abstract
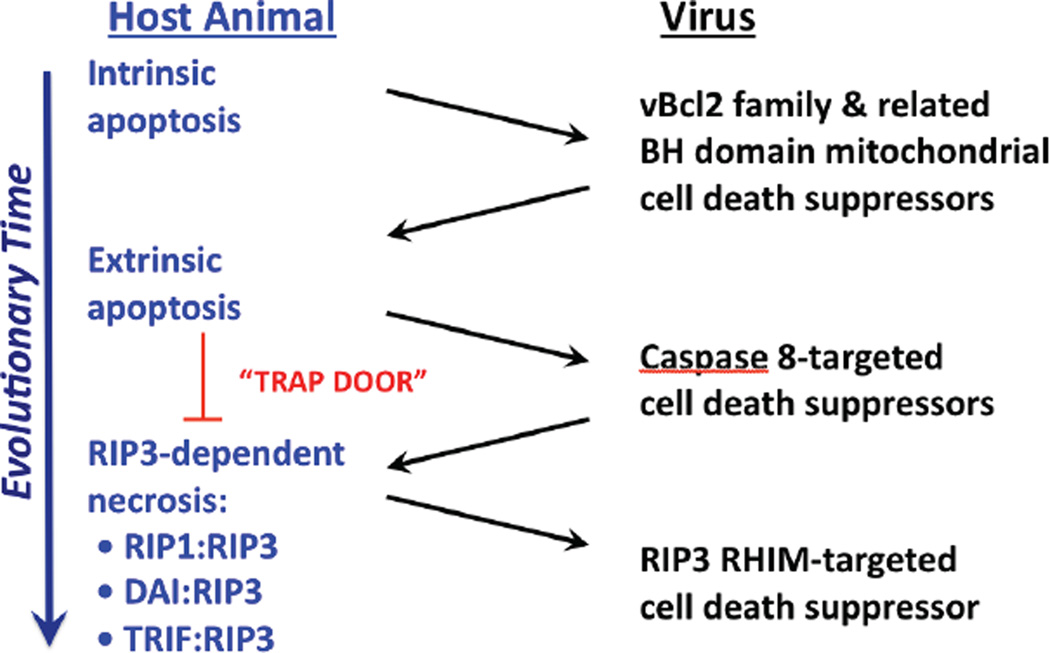
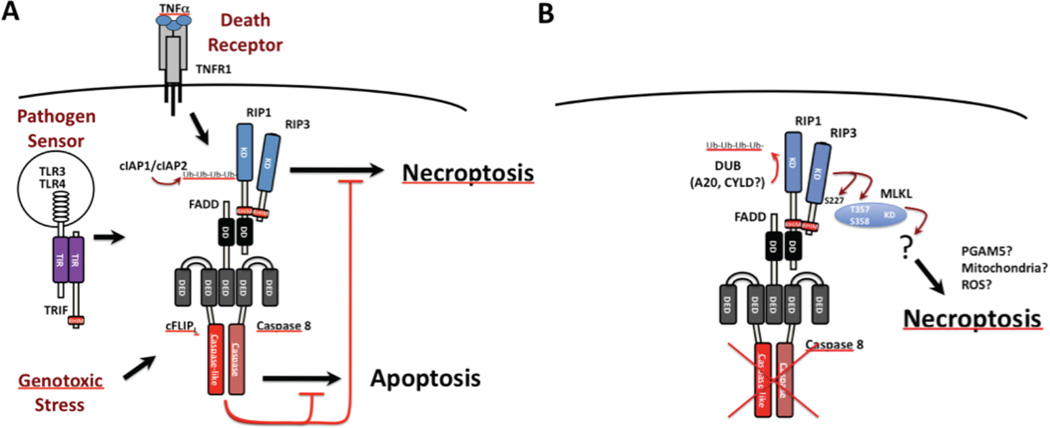
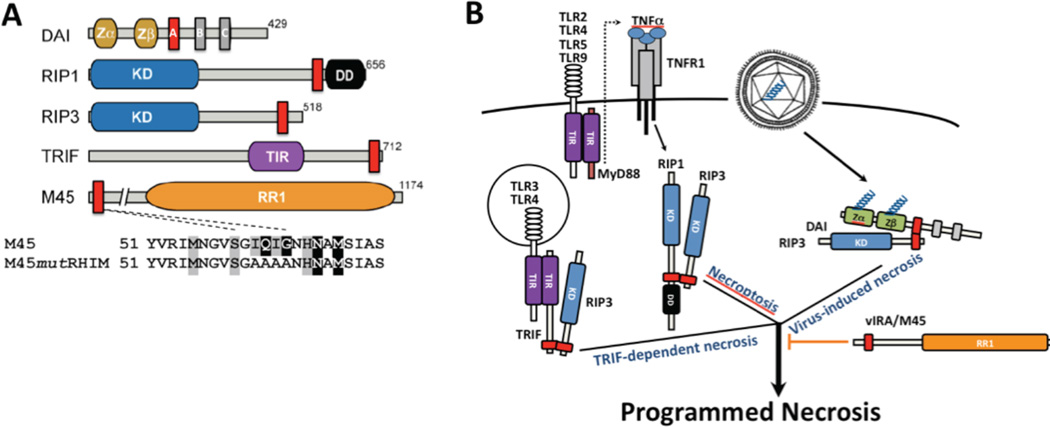
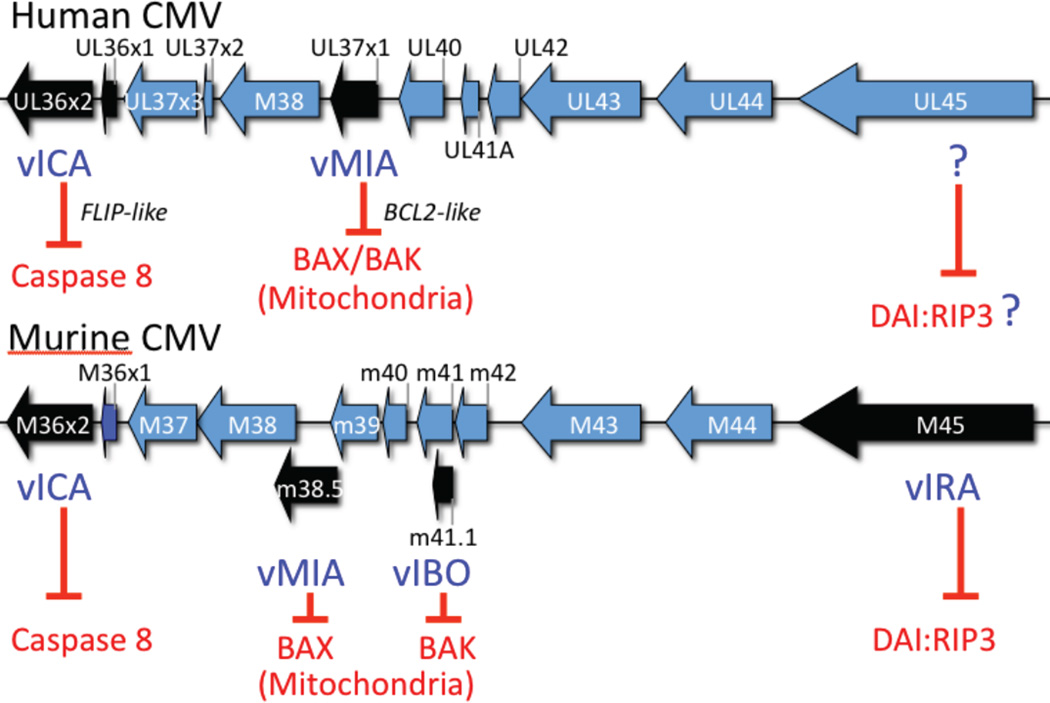
Apoptosis and programmed necrosis balance each other as alternate first line host defense pathways against which viruses have evolved countermeasures. Intrinsic apoptosis, the critical programmed cell death pathway that removes excess cells during embryonic development and tissue homeostasis, follows a caspase cascade triggered at mitochondria and modulated by virus-encoded anti-apoptotic B cell leukemia (BCL)2-like suppressors. Extrinsic apoptosis controlled by caspase 8 arose during evolution to trigger executioner caspases directly, circumventing viral suppressors of intrinsic (mitochondrial) apoptosis and providing the selective pressure for viruses to acquire caspase 8 suppressors. Programmed necrosis likely evolved most recently as a 'trap door' adaptation to extrinsic apoptosis. Receptor interacting protein (RIP)3 kinase (also called RIPK3) becomes active when either caspase 8 activity or polyubiquitylation of RIP1 is compromised. This evolutionary dialog implicates caspase 8 as a 'supersensor' alternatively activating and suppressing cell death pathways.
Copyright © 2013. Published by Elsevier B.V.
Figures
References
-
-
Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell. 2004;116:205–219. The classic review of intrinsic cell death reliance on BCL2 family members.
-
-
- Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev. 2007;87:99–163. - PubMed
-
- Everett H, McFadden G. Apoptosis: an innate immune response to virus infection. Trends Microbiol. 1999;7:160–165. - PubMed
-
- Benedict CA, Norris PS, Ware CF. To kill or be killed: viral evasion of apoptosis. Nat Immunol. 2002;3:1013–1018. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
