

Contribution of aldosterone to cardiovascular and renal inflammation and fibrosis
- PMID: 23774812
- PMCID: PMC3922409
- DOI: 10.1038/nrneph.2013.110
Contribution of aldosterone to cardiovascular and renal inflammation and fibrosis
Abstract
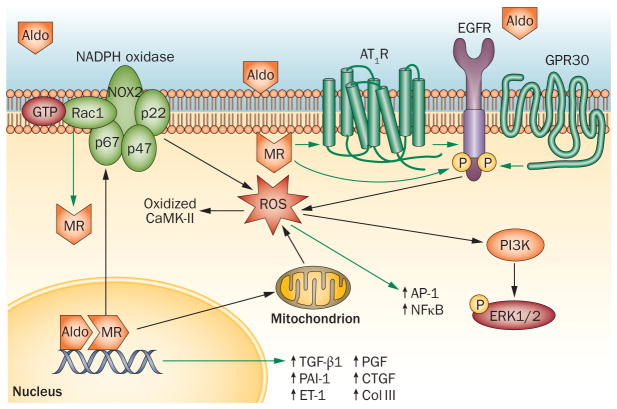
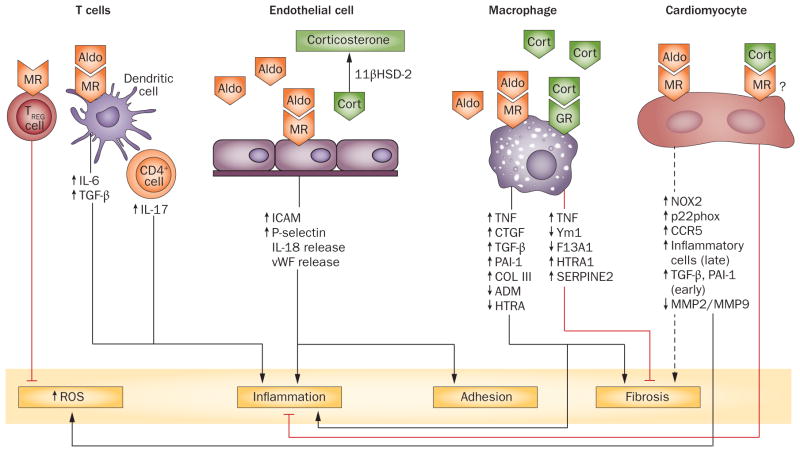
The steroid hormone aldosterone regulates sodium and potassium homeostasis. Aldosterone and activation of the mineralocorticoid receptor also causes inflammation and fibrosis of the heart, fibrosis and remodelling of blood vessels and tubulointerstitial fibrosis and glomerular injury in the kidney. Aldosterone and mineralocorticoid-receptor activation initiate an inflammatory response by increasing the generation of reactive oxygen species by nicotinamide adenine dinucleotide phosphate (NADPH) oxidase and mitochondria. High salt intake potentiates these effects, in part by activating the Rho family member Rac1, a regulatory subunit of reduced NADPH oxidase that activates the mineralocorticoid receptor. Studies in mice in which the mineralocorticoid receptor has been deleted from specific cell types suggest a key role for macrophages in promoting inflammation and fibrosis. Aldosterone can exert mineralocorticoid-receptor-independent effects via the angiotensin II receptor and via G-protein-coupled receptor 30. Mineralocorticoid-receptor antagonists are associated with decreased mortality in patients with heart disease and show promise in patients with kidney injury, but can elevate serum potassium concentration. Studies in rodents genetically deficient in aldosterone synthase or treated with a pharmacological aldosterone-synthase inhibitor are providing insight into the relative contribution of aldosterone compared with the contribution of mineralocorticoid-receptor activation in inflammation, fibrosis, and injury. Aldosterone-synthase inhibitors are under development in humans.
Conflict of interest statement
The author declares associations with the following companies: Novartis, Shire Pharmaceuticals. See the article online for full details of the relationships.
Figures
References
-
- Náray-Fejes-Tóth A, Canessa C, Cleaveland ES, Aldrich G, Fejes-Tóth G. sgk is an aldosterone-induced kinase in the renal collecting duct Effects on epithelial na+ channels. J Biol Chem. 1999;274:16973–16978. - PubMed
-
- Snyder PM, Olson DR, Thomas BC. Serum and glucocorticoid-regulated kinase modulates Nedd4-2-mediated inhibition of the epithelial Na+ channel. J Biol Chem. 2002;277:5–8. - PubMed
-
- Bhalla V, Soundararajan R, Pao AC, Li H, Pearce D. Disinhibitory pathways for control of sodium transport: regulation of ENaC by SGK1 and GILZ. Am J Physiol Renal Physiol. 2006;291:F714–F721. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
