

Crystal structure of prothrombin reveals conformational flexibility and mechanism of activation
- PMID: 23775088
- PMCID: PMC3829358
- DOI: 10.1074/jbc.M113.466946
Crystal structure of prothrombin reveals conformational flexibility and mechanism of activation
Abstract
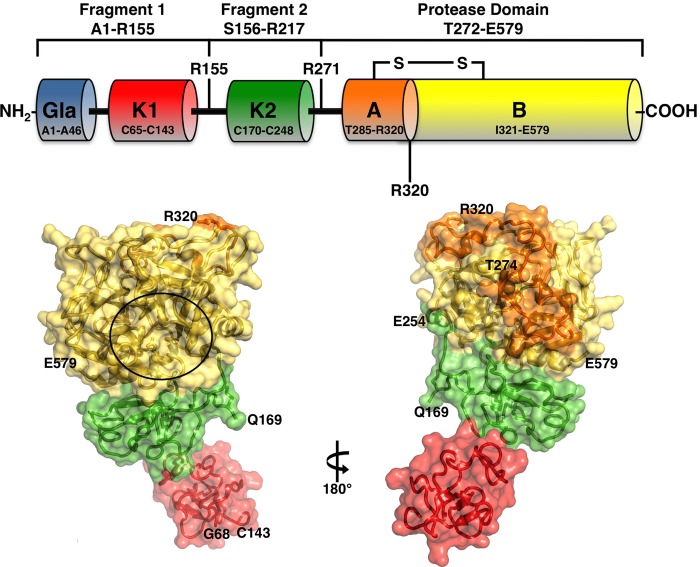
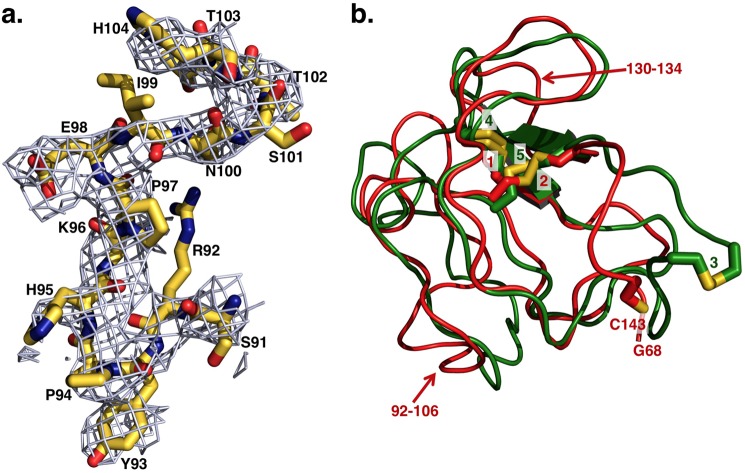
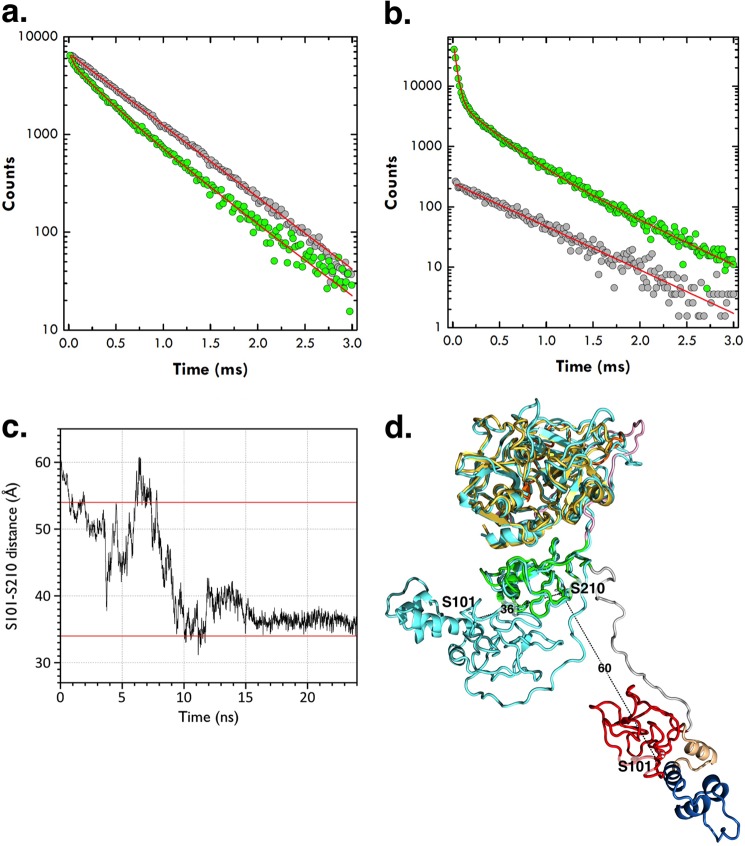
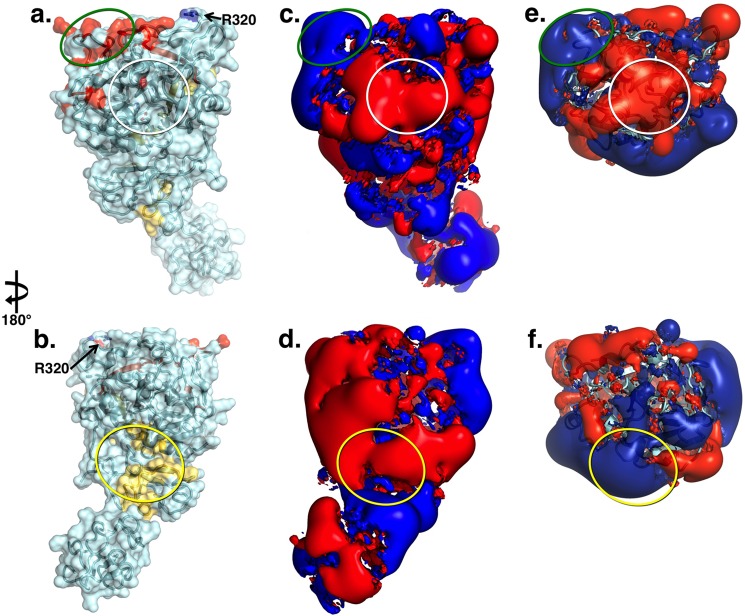
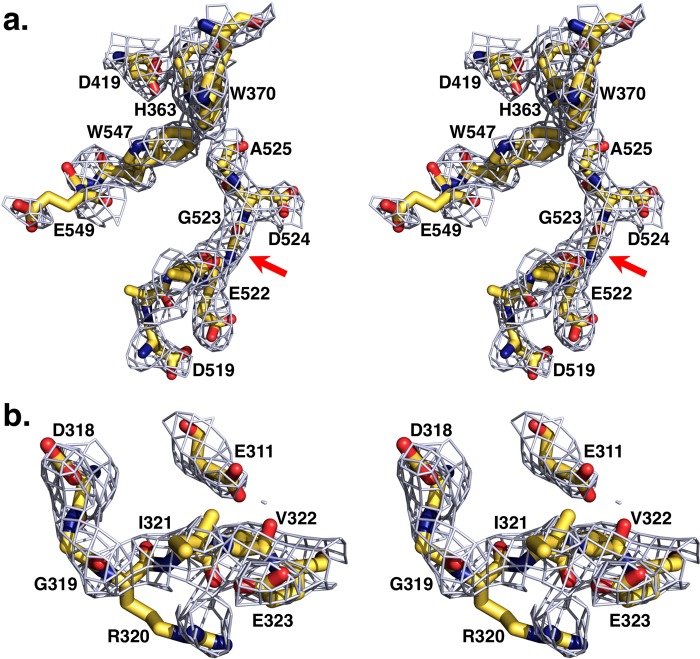
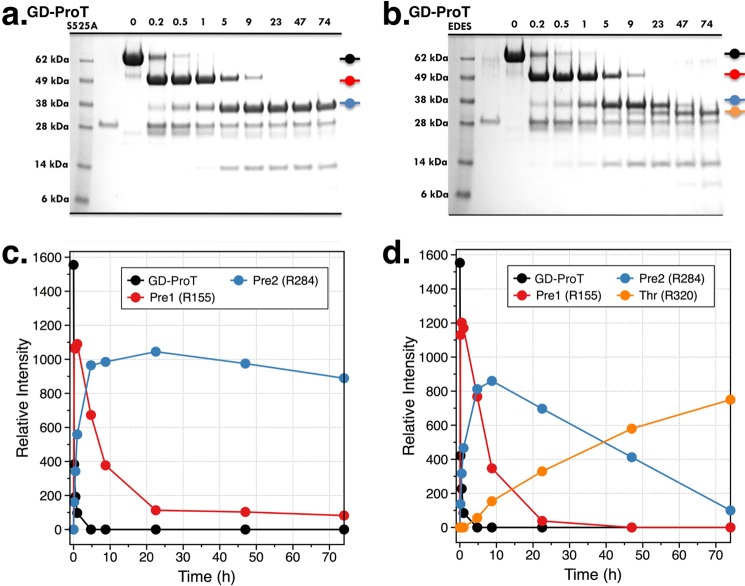
The zymogen prothrombin is composed of fragment 1 containing a Gla domain and kringle-1, fragment 2 containing kringle-2, and a protease domain containing A and B chains. The prothrombinase complex assembled on the surface of platelets converts prothrombin to thrombin by cleaving at Arg-271 and Arg-320. The three-dimensional architecture of prothrombin and the molecular basis of its activation remain elusive. Here we report the first x-ray crystal structure of prothrombin as a Gla-domainless construct carrying an Ala replacement of the catalytic Ser-525. Prothrombin features a conformation 80 Å long, with fragment 1 positioned at a 36° angle relative to the main axis of fragment 2 coaxial to the protease domain. High flexibility of the linker connecting the two kringles suggests multiple arrangements for kringle-1 relative to the rest of the prothrombin molecule. Luminescence resonance energy transfer measurements detect two distinct conformations of prothrombin in solution, in a 3:2 ratio, with the distance between the two kringles either fully extended (54 ± 2 Å) or partially collapsed (≤34 Å) as seen in the crystal structure. A molecular mechanism of prothrombin activation emerges from the structure. Of the two sites of cleavage, Arg-271 is located in a disordered region connecting kringle-2 to the A chain, but Arg-320 is well defined within the activation domain and is not accessible to proteolysis in solution. Burial of Arg-320 prevents prothrombin autoactivation and directs prothrombinase to cleave at Arg-271 first. Reversal of the local electrostatic potential then redirects prothrombinase toward Arg-320, leading to thrombin generation via the prethrombin-2 intermediate.
Keywords: Coagulation factors; Enzyme mutation; Enzyme structure; Structural biology; Thrombin; Zymogen.
Figures
References
-
- Schmidt A. (1872) Neue Untersuchungen ueber die Fasserstoffesgerinnung. Pflüger Archiv. 6, 413–538
-
- Butenas S., van't Veer C., Mann K. G. (1999) “Normal” thrombin generation. Blood 94, 2169–2178 - PubMed
-
- Rosing J., Tans G., Govers-Riemslag J. W., Zwaal R. F., Hemker H. C. (1980) The role of phospholipids and factor Va in the prothrombinase complex. J. Biol. Chem. 255, 274–283 - PubMed
-
- Kotkow K. J., Furie B., Furie B. C. (1993) The interaction of prothrombin with phospholipid membranes is independent of either kringle domain. J. Biol. Chem. 268, 15633–15639 - PubMed
-
- Huang M., Rigby A. C., Morelli X., Grant M. A., Huang G., Furie B., Seaton B., Furie B. C. (2003) Structural basis of membrane binding by Gla domains of vitamin K-dependent proteins. Nat. Struct. Biol. 10, 751–756 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
