

Inhibition of fast axonal transport by pathogenic SOD1 involves activation of p38 MAP kinase
- PMID: 23776455
- PMCID: PMC3680447
- DOI: 10.1371/journal.pone.0065235
Inhibition of fast axonal transport by pathogenic SOD1 involves activation of p38 MAP kinase
Abstract
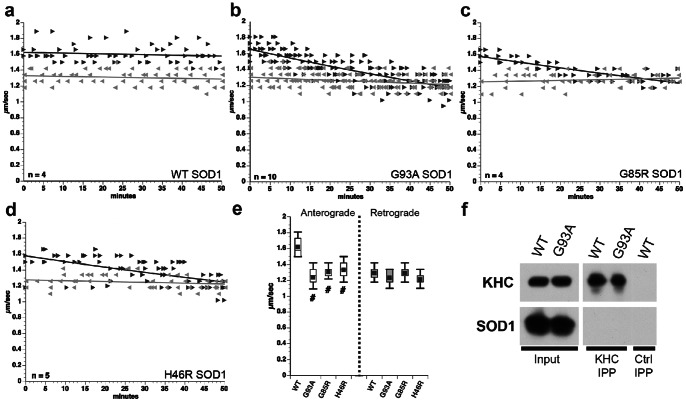
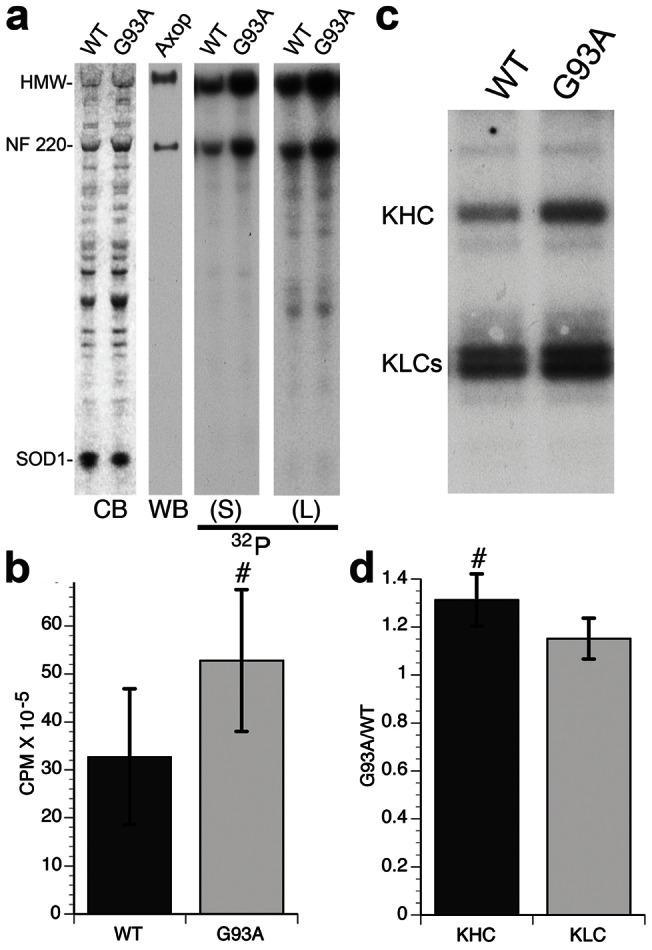
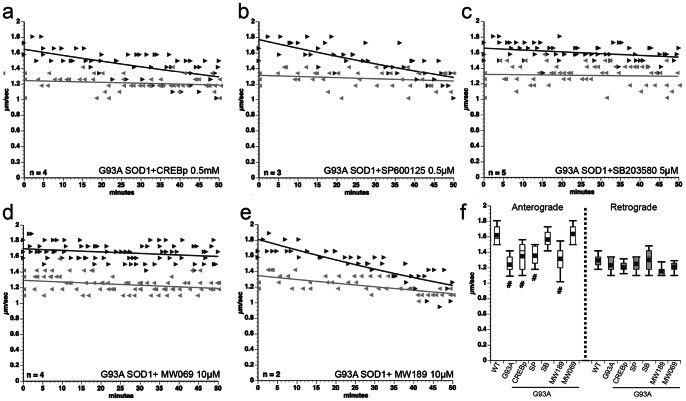
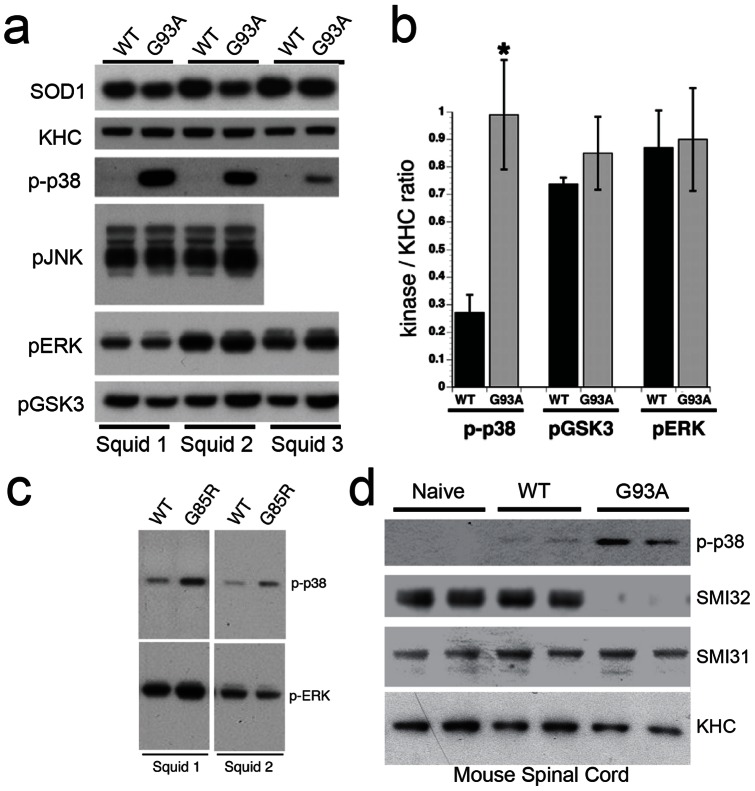
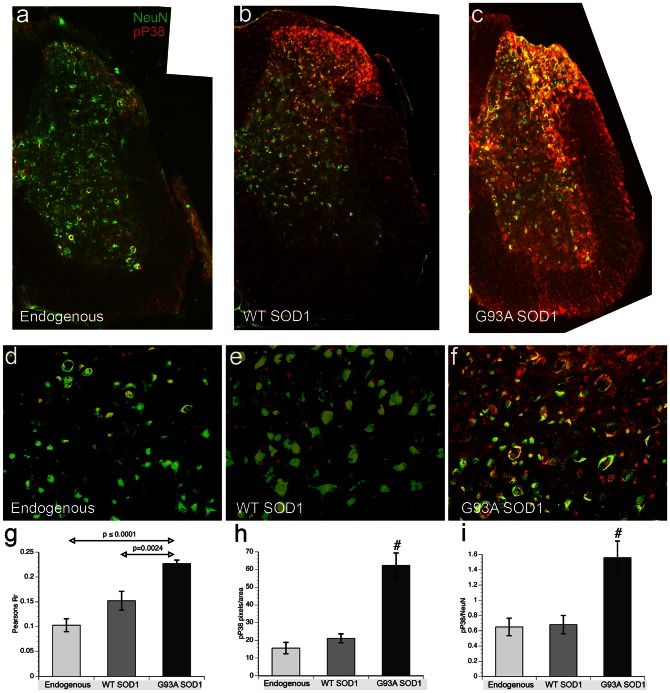
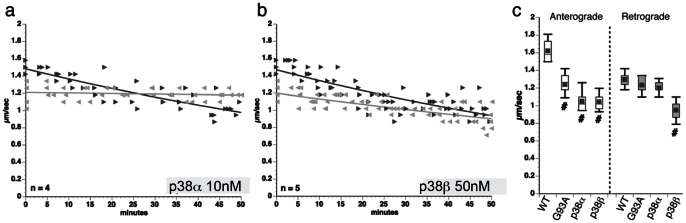
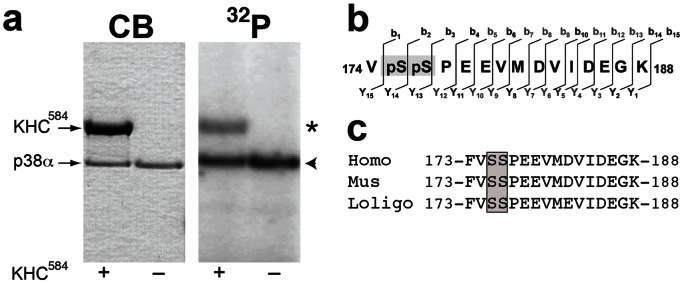
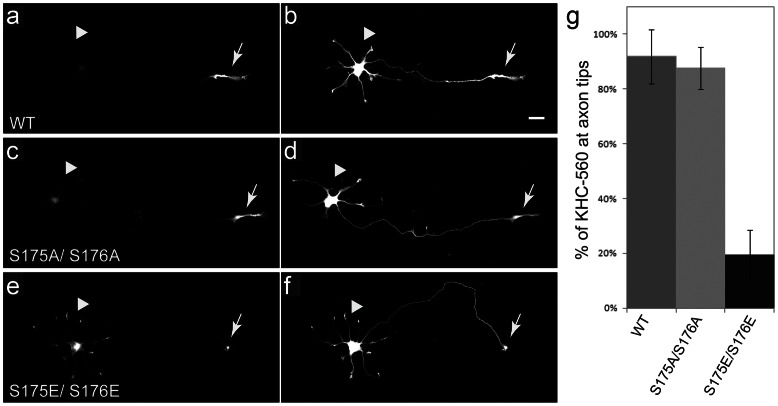
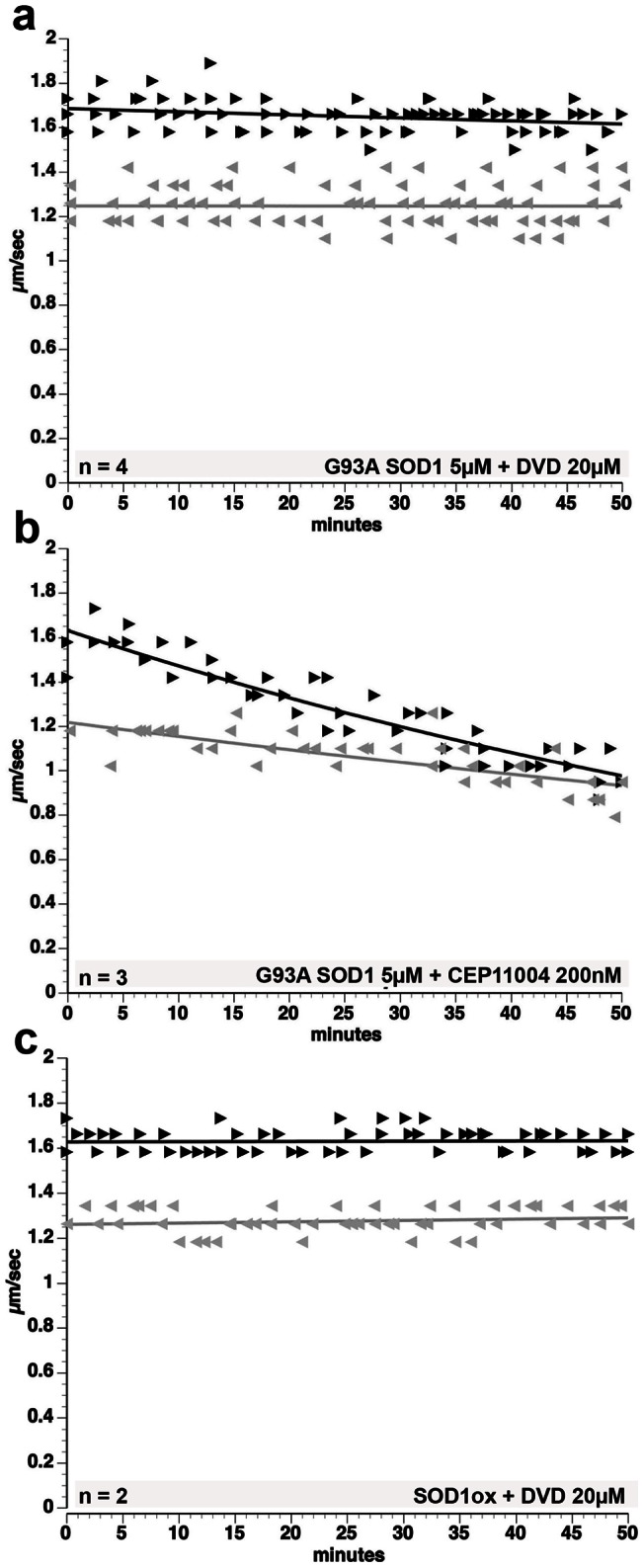
Dying-back degeneration of motor neuron axons represents an established feature of familial amyotrophic lateral sclerosis (FALS) associated with superoxide dismutase 1 (SOD1) mutations, but axon-autonomous effects of pathogenic SOD1 remained undefined. Characteristics of motor neurons affected in FALS include abnormal kinase activation, aberrant neurofilament phosphorylation, and fast axonal transport (FAT) deficits, but functional relationships among these pathogenic events were unclear. Experiments in isolated squid axoplasm reveal that FALS-related SOD1 mutant polypeptides inhibit FAT through a mechanism involving a p38 mitogen activated protein kinase pathway. Mutant SOD1 activated neuronal p38 in mouse spinal cord, neuroblastoma cells and squid axoplasm. Active p38 MAP kinase phosphorylated kinesin-1, and this phosphorylation event inhibited kinesin-1. Finally, vesicle motility assays revealed previously unrecognized, isoform-specific effects of p38 on FAT. Axon-autonomous activation of the p38 pathway represents a novel gain of toxic function for FALS-linked SOD1 proteins consistent with the dying-back pattern of neurodegeneration characteristic of ALS.
Conflict of interest statement
Figures
References
-
- Bruijn LI, Miller TM, Cleveland DW (2004) Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annu Rev Neurosci 27: 723–749. - PubMed
-
- Dion PA, Daoud H, Rouleau GA (2009) Genetics of motor neuron disorders: new insights into pathogenic mechanisms. Nat Rev Genet 10: 769–782. - PubMed
-
- Pasinelli P, Brown RH (2006) Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat Rev Neurosci 7: 710–723. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
- R01NS067206/NS/NINDS NIH HHS/United States
- RC2 NS070342/NS/NINDS NIH HHS/United States
- R01AG031311/AG/NIA NIH HHS/United States
- NS41170/NS/NINDS NIH HHS/United States
- R01NS055951/NS/NINDS NIH HHS/United States
- 52006287/HHMI/Howard Hughes Medical Institute/United States
- R01 NS050557/NS/NINDS NIH HHS/United States
- R01 NS067206/NS/NINDS NIH HHS/United States
- NS23320/NS/NINDS NIH HHS/United States
- 1RC2NS070342/NS/NINDS NIH HHS/United States
- R56 NS023868/NS/NINDS NIH HHS/United States
- R01NS050557/NS/NINDS NIH HHS/United States
- R01 NS055951/NS/NINDS NIH HHS/United States
- R01 AG031311/AG/NIA NIH HHS/United States
- R01 NS023320/NS/NINDS NIH HHS/United States
- U01NS05225/NS/NINDS NIH HHS/United States
- R01 NS041170/NS/NINDS NIH HHS/United States
- U01 AG043415/AG/NIA NIH HHS/United States
- R01NS44170/NS/NINDS NIH HHS/United States
- RC1 NS068391/NS/NINDS NIH HHS/United States
- NS23868/NS/NINDS NIH HHS/United States
- MH066179/MH/NIMH NIH HHS/United States
- R01 NS044170/NS/NINDS NIH HHS/United States
- 1RC1NS068391/NS/NINDS NIH HHS/United States
- R01 MH066179/MH/NIMH NIH HHS/United States
- NS066942A/NS/NINDS NIH HHS/United States
- R01 NS023868/NS/NINDS NIH HHS/United States
- R01 NS066942/NS/NINDS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
