

Functional assessment of population and tumor-associated APE1 protein variants
- PMID: 23776569
- PMCID: PMC3679070
- DOI: 10.1371/journal.pone.0065922
Functional assessment of population and tumor-associated APE1 protein variants
Abstract
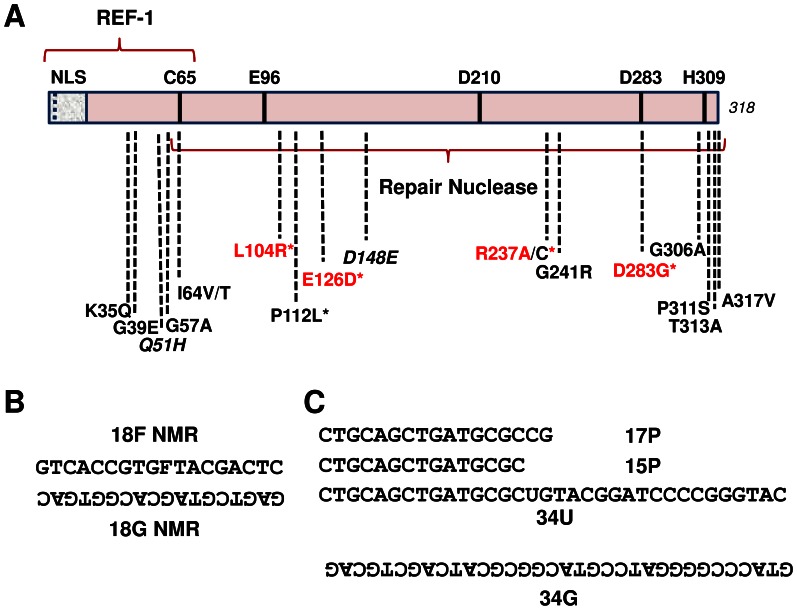
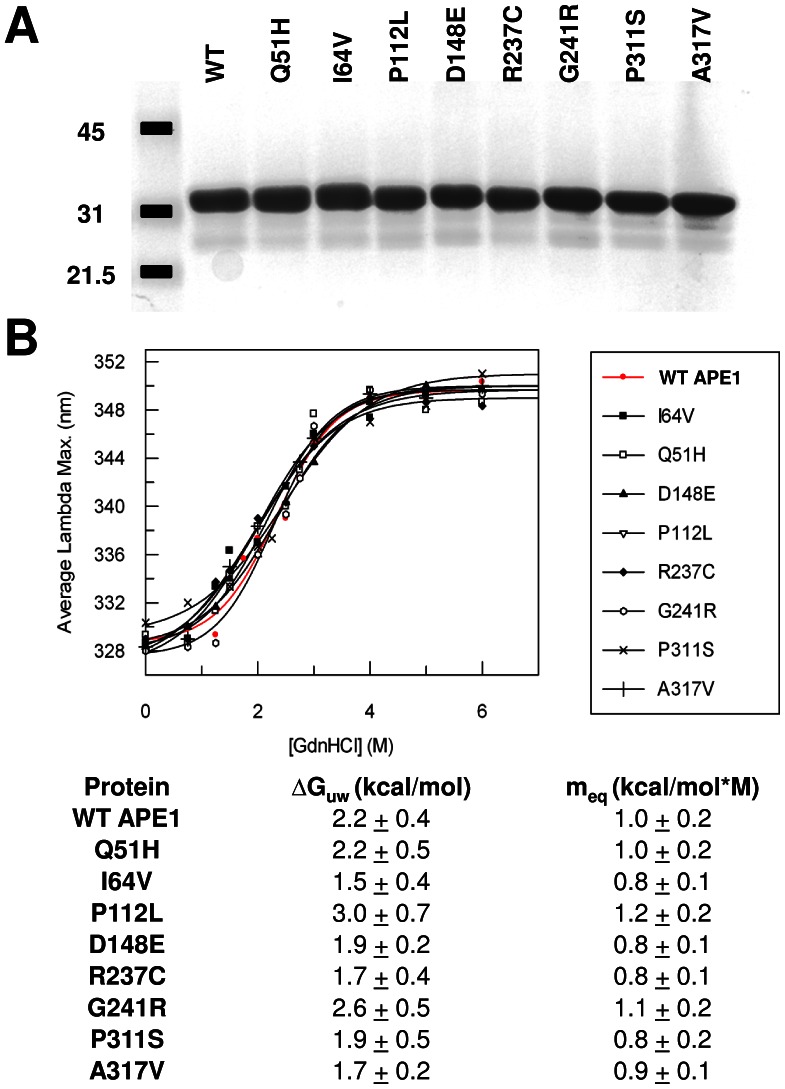
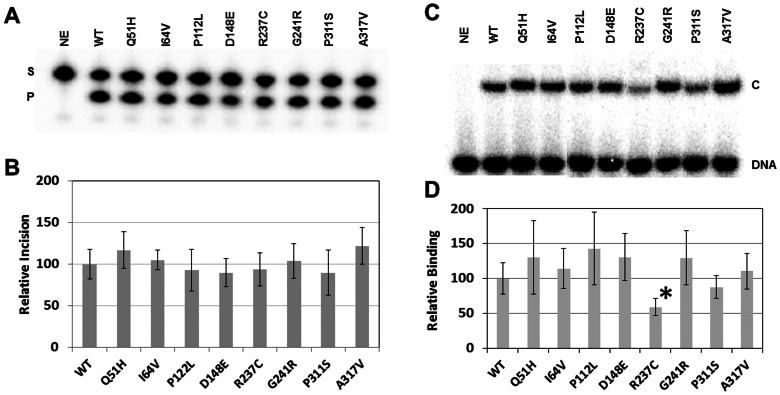
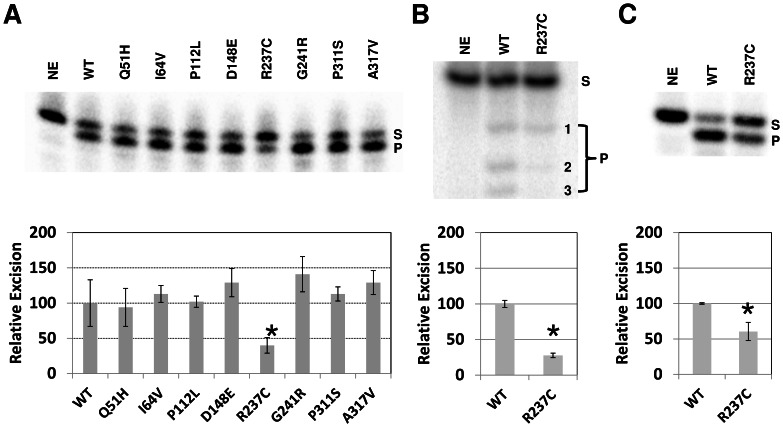
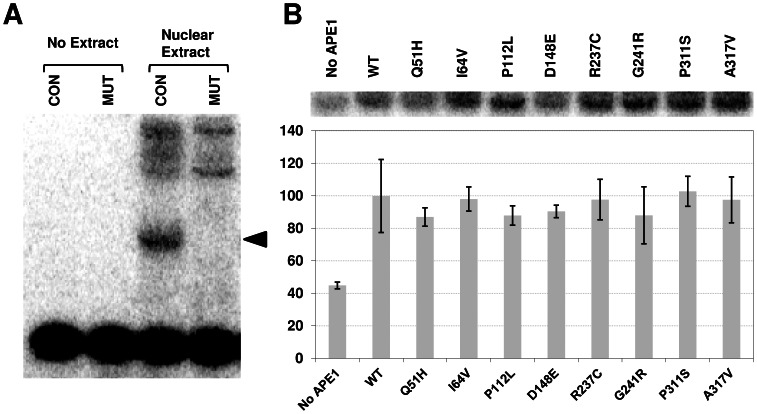
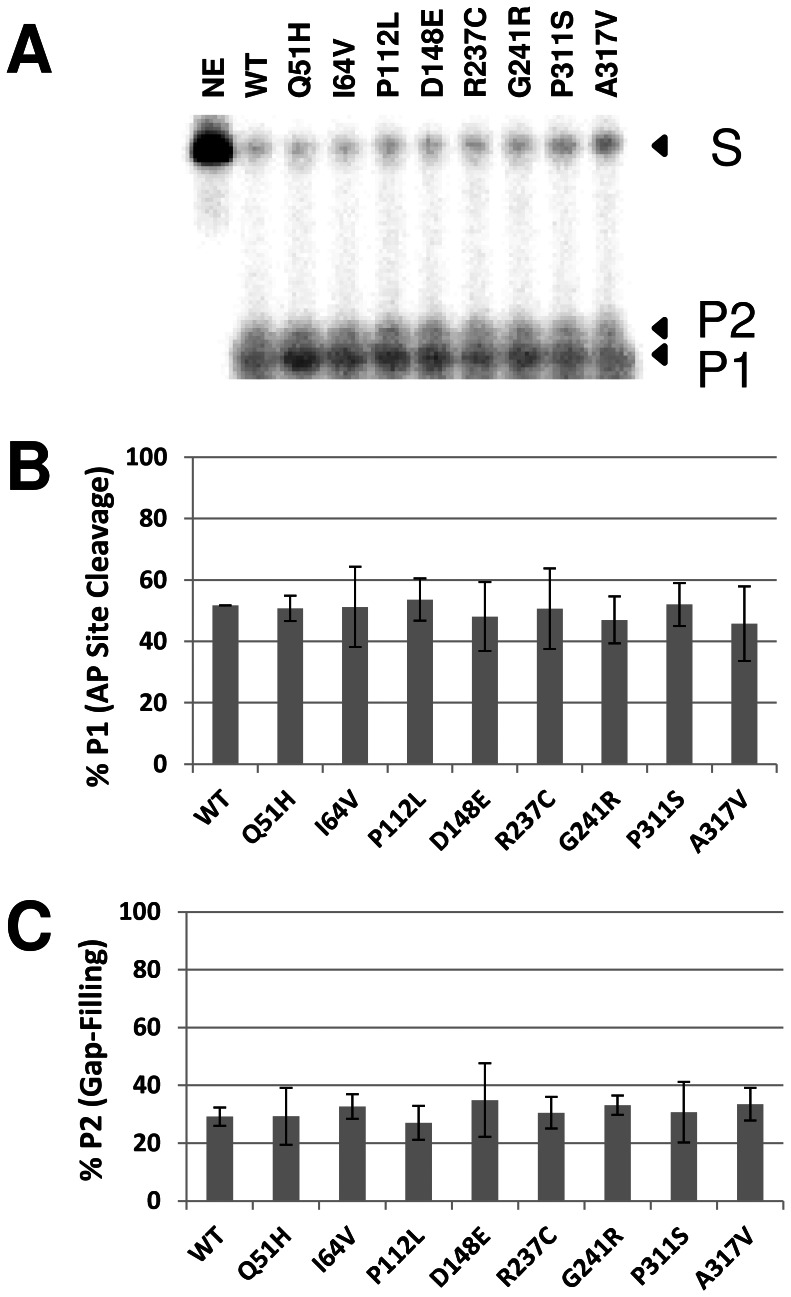
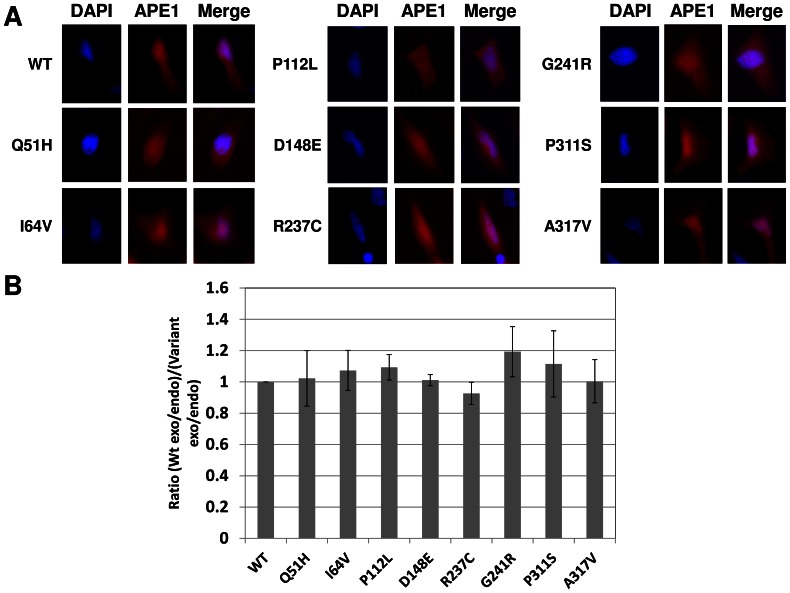
Apurinic/apyrimidinic endonuclease 1 (APE1) is the predominant AP site repair enzyme in mammals. APE1 also maintains 3'-5' exonuclease and 3'-repair activities, and regulates transcription factor DNA binding through its REF-1 function. Since complete or severe APE1 deficiency leads to embryonic lethality and cell death, it has been hypothesized that APE1 protein variants with slightly impaired function will contribute to disease etiology. Our data indicate that except for the endometrial cancer-associated APE1 variant R237C, the polymorphic variants Q51H, I64V and D148E, the rare population variants G241R, P311S and A317V, and the tumor-associated variant P112L exhibit normal thermodynamic stability of protein folding; abasic endonuclease, 3'-5' exonuclease and REF-1 activities; coordination during the early steps of base excision repair; and intracellular distribution when expressed exogenously in HeLa cells. The R237C mutant displayed reduced AP-DNA complex stability, 3'-5' exonuclease activity and 3'-damage processing. Re-sequencing of the exonic regions of APE1 uncovered no novel amino acid substitutions in the 60 cancer cell lines of the NCI-60 panel, or in HeLa or T98G cancer cell lines; only the common D148E and Q51H variants were observed. Our results indicate that APE1 missense mutations are seemingly rare and that the cancer-associated R237C variant may represent a reduced-function susceptibility allele.
Conflict of interest statement
Figures
References
-
- Lindahl T (1993) Instability and decay of the primary structure of DNA. Nature 362: 709–715. - PubMed
-
- Loeb LA, Preston BD (1986) Mutagenesis by apurinic/apyrimidinic sites. Ann Rev Genet 20: 201–230. - PubMed
-
- Fortini P, Dogliotti E (2007) Base damage and single-strand break repair: mechanisms and functional significance of short- and long-patch repair subpathways. DNA Repair (Amst) 6: 398–409 S1568–7864(06)00317-X [pii];10.1016/j.dnarep.2006.10.008 [doi] - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
