

A MEN1 syndrome with a paraganglioma
- PMID: 23778871
- PMCID: PMC3895646
- DOI: 10.1038/ejhg.2013.128
A MEN1 syndrome with a paraganglioma
Abstract
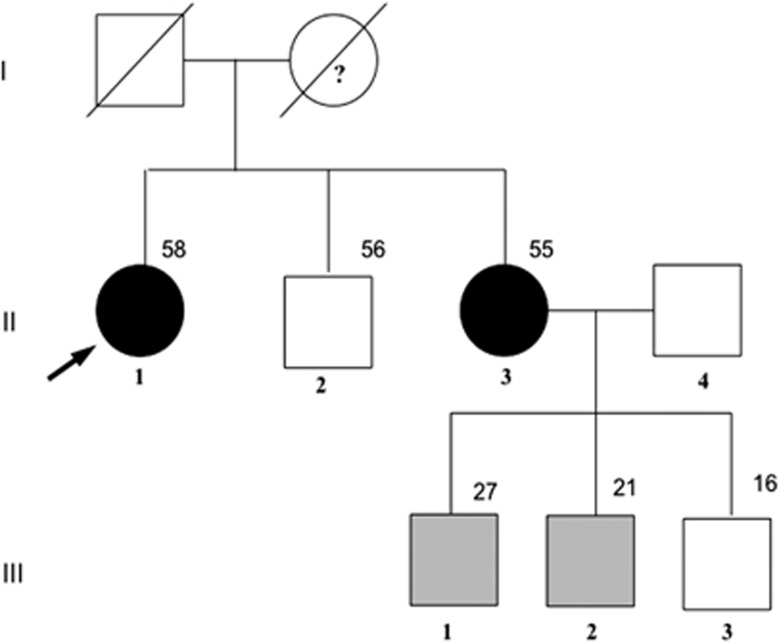
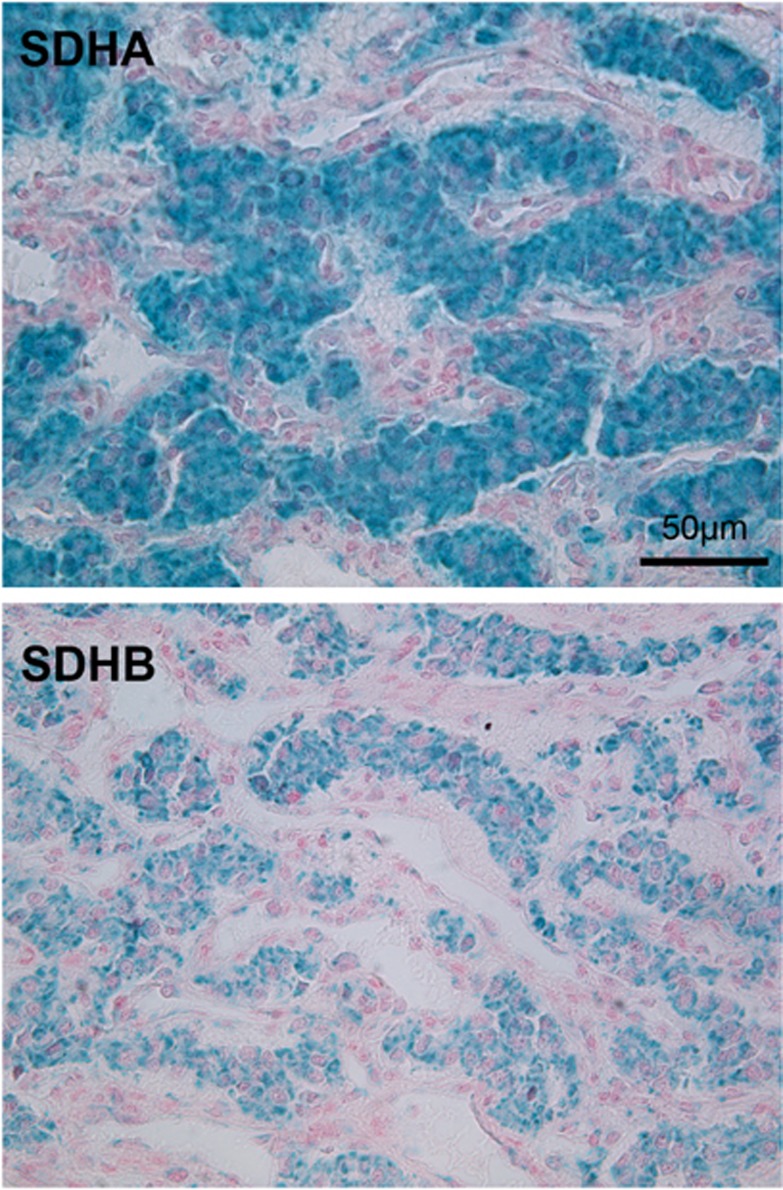
Germline mutations of the MEN1 gene cause multiple endocrine neoplasia type 1 (MEN1), an autosomal dominant disorder characterized by tumors of the parathyroids, the pancreas, and the anterior pituitary. Paraganglioma (PGL) is a rare endocrine tumor, which can be sporadic or genetically determined. To date, PGL has never been reported as a feature of MEN1.We report here a patient presenting three features of MEN1 syndrome (hyperparathyroidism, pancreatic neuroendocrine tumor, and adrenocortical adenoma) associated with PGL. Genetic analysis of MEN1 gene revealed a new missense mutation in exon 5 (AGGAAG), causing the substitution of arginine by lysine at codon 275. Screening for other genetic disorders (SDHx, TMEM127, MAX, CDKN1B) causing PGL was negative. Immunohistochemical analyses showed normal levels of succinate dehydrogenase (SDH)A and SDHB in the PGL. The proband's sister, bearing the mutation, had primary hyperparathyroidism. It was the first typical MEN1 syndrome reported with an extra-adrenal PGL.
Figures
Similar articles
-
Primary Renal Paragangliomas and Renal Neoplasia Associated with Pheochromocytoma/Paraganglioma: Analysis of von Hippel-Lindau (VHL), Succinate Dehydrogenase (SDHX) and Transmembrane Protein 127 (TMEM127).Endocr Pathol. 2017 Sep;28(3):253-268. doi: 10.1007/s12022-017-9489-0. Endocr Pathol. 2017. PMID: 28646318
-
Pediatric paraganglioma: an early manifestation of an adult disease secondary to germline mutations.Pediatr Blood Cancer. 2006 Nov;47(6):785-9. doi: 10.1002/pbc.20680. Pediatr Blood Cancer. 2006. PMID: 16304664
-
Novel SDHB and TMEM127 Mutations in Patients with Pheochromocytoma/Paraganglioma Syndrome.Pathol Oncol Res. 2016 Oct;22(4):673-9. doi: 10.1007/s12253-016-0050-0. Epub 2016 Mar 9. Pathol Oncol Res. 2016. PMID: 26960314
-
[Clinical symptoms, diagnosis and treatment of multiple endocrine neoplasia type 1. Results of genetic screening in Hungarian patients].Orv Hetil. 2005 Oct 23;146(43):2191-7. Orv Hetil. 2005. PMID: 16323565 Review. Hungarian.
-
Multiple endocrine neoplasia type 1.Orphanet J Rare Dis. 2006 Oct 2;1:38. doi: 10.1186/1750-1172-1-38. Orphanet J Rare Dis. 2006. PMID: 17014705 Free PMC article. Review.
Cited by
-
Coexistence of DIPNECH and carotid body paraganglioma: is it just a coincidence?Endocrinol Diabetes Metab Case Rep. 2020 May 13;2020:EDM19-0141. doi: 10.1530/EDM-19-0141. Online ahead of print. Endocrinol Diabetes Metab Case Rep. 2020. PMID: 32408270 Free PMC article.
-
Rare germline mutations identified by targeted next-generation sequencing of susceptibility genes in pheochromocytoma and paraganglioma.J Clin Endocrinol Metab. 2014 Jul;99(7):E1352-60. doi: 10.1210/jc.2013-4375. Epub 2014 Apr 2. J Clin Endocrinol Metab. 2014. PMID: 24694336 Free PMC article.
-
Molecular markers of paragangliomas/pheochromocytomas.Oncotarget. 2017 Apr 11;8(15):25756-25782. doi: 10.18632/oncotarget.15201. Oncotarget. 2017. PMID: 28187001 Free PMC article. Review.
-
Non-Susceptibility Gene Variants in Head and Neck Paragangliomas.Int J Mol Sci. 2024 Nov 27;25(23):12762. doi: 10.3390/ijms252312762. Int J Mol Sci. 2024. PMID: 39684472 Free PMC article.
-
Genetic Landscape and Clinical Manifestations of Multiple Endocrine Neoplasia Type 1 in a Korean Cohort: A Multicenter Retrospective Analysis.Endocrinol Metab (Seoul). 2024 Dec;39(6):956-964. doi: 10.3803/EnM.2024.2008. Epub 2024 Nov 18. Endocrinol Metab (Seoul). 2024. PMID: 39552147 Free PMC article.
References
-
- Larsson C, Skogseid B, Oberg K, Nakamura Y, Nordenskjöld M. Multiple endocrine neoplasia type 1 gene maps to chromosome 11 and is lost in insulinoma. Nature. 1988;332:85–87. - PubMed
-
- Agarwal SK, Kester MB, Debelenko LV, et al. Germline mutations of the MEN1 gene in familial multiple endocrine neoplasia type 1 and related states. Hum Mol Genet. 1997;6:1169–1175. - PubMed
-
- Knudson AG, Jr, Strong LC, Anderson DE. Heredity and cancer in man. Prog Med Genet. 1973;9:113–158. - PubMed
-
- Welander J, Söderkvist P, Gimm O. Genetics and clinical characteristics of hereditary pheochromocytomas and paragangliomas. Endocr Relat Cancer. 2011;18:R253–R276. - PubMed
-
- Gimenez-Roqueplo AP, Dahia PL, Robledo M. An update on the genetics of paraganglioma, pheochromocytoma, and associated hereditary syndromes. Horm Metab Res. 2012;44:328–333. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
