

Cross-sectional observational study of 208 patients with non-classical urea cycle disorders
- PMID: 23780642
- PMCID: PMC3889631
- DOI: 10.1007/s10545-013-9624-0
Cross-sectional observational study of 208 patients with non-classical urea cycle disorders
Abstract
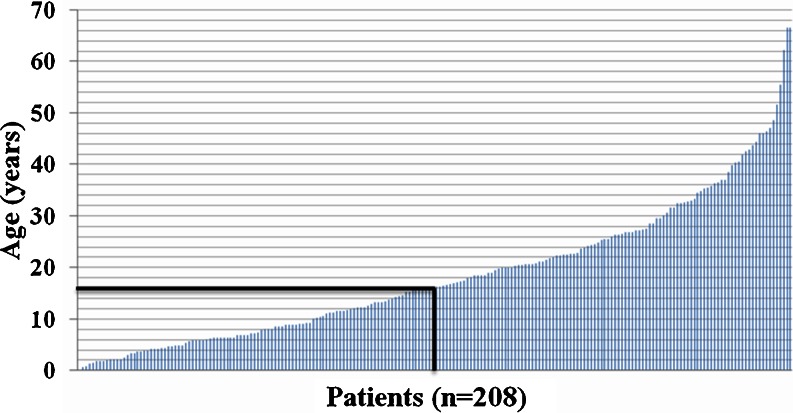
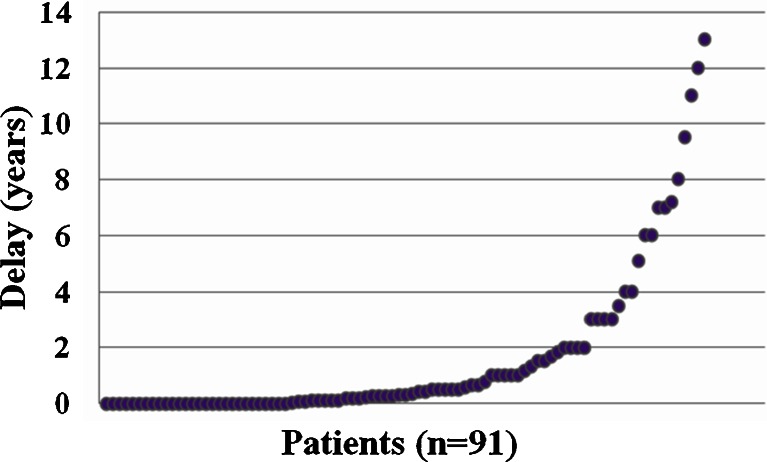
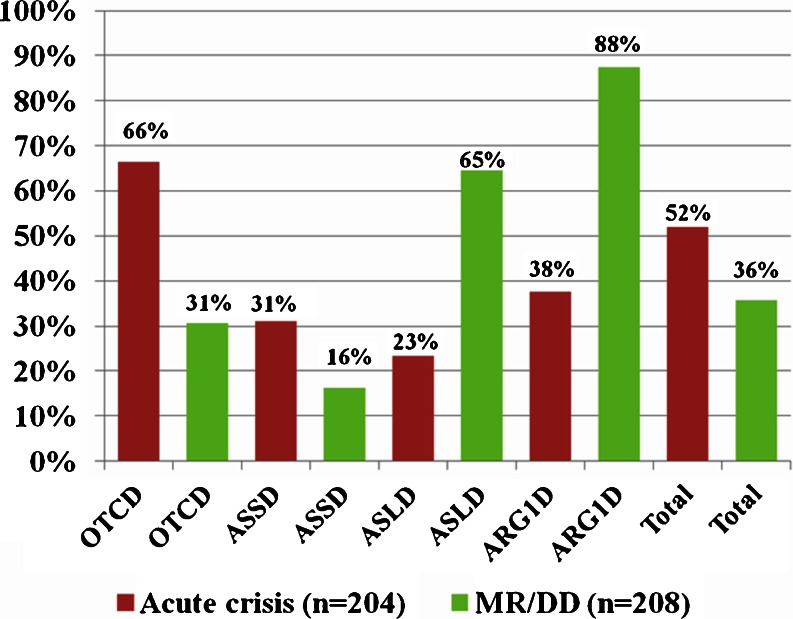
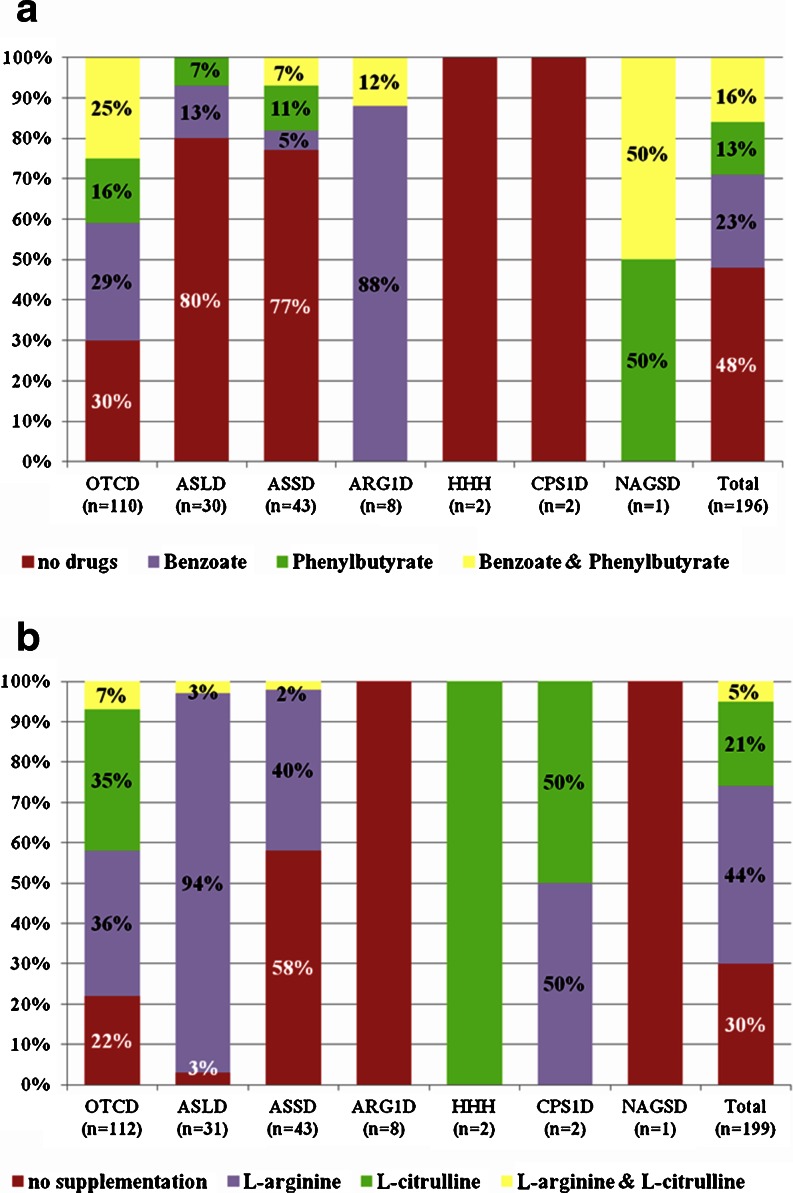
Urea cycle disorders (UCDs) are inherited disorders of ammonia detoxification often regarded as mainly of relevance to pediatricians. Based on an increasing number of case studies it has become obvious that a significant number of UCD patients are affected by their disease in a non-classical way: presenting outside the newborn period, following a mild course, presenting with unusual clinical features, or asymptomatic patients with only biochemical signs of a UCD. These patients are surviving into adolescence and adulthood, rendering this group of diseases clinically relevant to adult physicians as well as pediatricians. In preparation for an international workshop we collected data on all patients with non-classical UCDs treated by the participants in 20 European metabolic centres. Information was collected on a cohort of 208 patients 50% of which were ≥ 16 years old. The largest subgroup (121 patients) had X-linked ornithine transcarbamylase deficiency (OTCD) of whom 83 were female and 29% of these were asymptomatic. In index patients, there was a mean delay from first symptoms to diagnosis of 1.6 years. Cognitive impairment was present in 36% of all patients including female OTCD patients (in 31%) and those 41 patients identified presymptomatically following positive newborn screening (in 12%). In conclusion, UCD patients with non-classical clinical presentations require the interest and care of adult physicians and have a high risk of neurological complications. To improve the outcome of UCDs, a greater awareness by health professionals of the importance of hyperammonemia and UCDs, and ultimately avoidance of the still long delay to correctly diagnose the patients, is crucial.
Figures
References
-
- Brusilow S, Horwich A. Urea cycle enzymes. In: Scriver C, Beaudet A, Sly W, Valle D, editors. The metabolic & molecular bases of inherited disease. 8. New York: McGraw-Hill; 2001. pp. 1909–1963.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
