

Impact of bioinformatic procedures in the development and translation of high-throughput molecular classifiers in oncology
- PMID: 23780890
- PMCID: PMC3745509
- DOI: 10.1158/1078-0432.CCR-12-3937
Impact of bioinformatic procedures in the development and translation of high-throughput molecular classifiers in oncology
Abstract
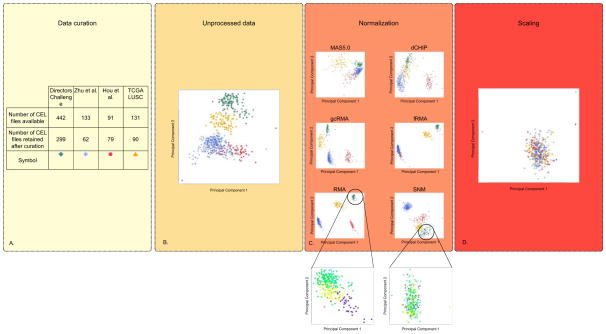
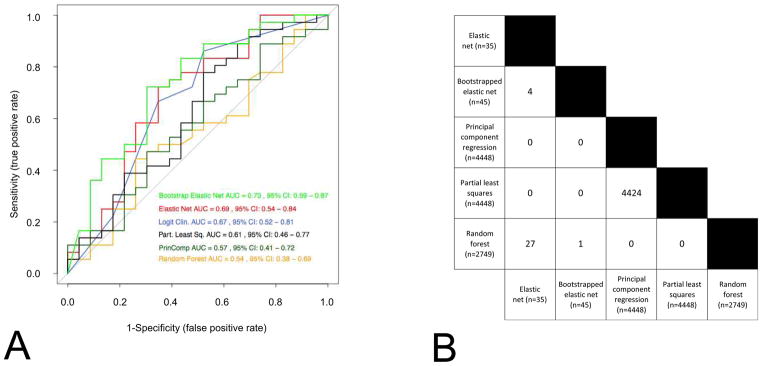
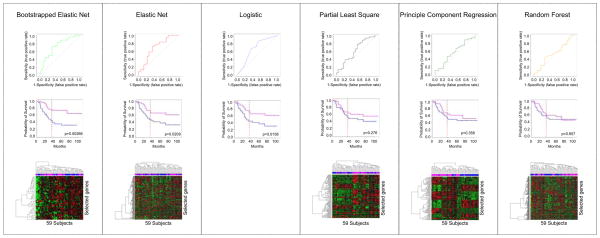
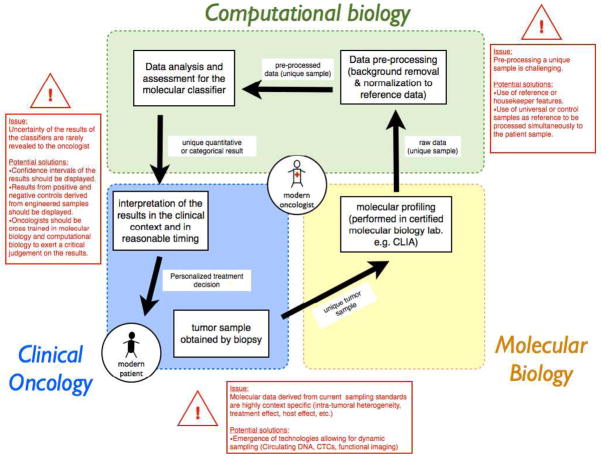
The progressive introduction of high-throughput molecular techniques in the clinic allows for the extensive and systematic exploration of multiple biologic layers of tumors. Molecular profiles and classifiers generated from these assays represent the foundation of what the National Academy describes as the future of "precision medicine". However, the analysis of such complex data requires the implementation of sophisticated bioinformatic and statistical procedures. It is critical that oncology practitioners be aware of the advantages and limitations of the methods used to generate classifiers to usher them into the clinic. This article uses publicly available expression data from patients with non-small cell lung cancer to first illustrate the challenges of experimental design and preprocessing of data before clinical application and highlights the challenges of high-dimensional statistical analysis. It provides a roadmap for the translation of such classifiers to clinical practice and makes key recommendations for good practice.
©2013 AACR.
Conflict of interest statement
Figures
References
-
- National Research Council of the National Academies. Toward Precision Medicine: Building a Knowledge Network for Biomedical Research and a New Taxonomy of Disease. 2011. - PubMed
-
- Ferté C, André F, Soria J-C. Molecular circuits of solid tumors: prognostic and predictive tools for bedside use. Nat Rev Clin Oncol. 2010;7:367–80. - PubMed
-
- Koscielny S. Why most gene expression signatures of tumors have not been useful in the clinic. Sci Transl Med. 2010;2:14ps2. - PubMed
-
- Mukherjee S, Tamayo P, Rogers S, Rifkin R, Engle A, Campbell C, et al. Estimating dataset size requirements for classifying DNA microarray data. J Comput Biol. 2003;10:119–42. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
