

Gangliosides and gangliosidoses: principles of molecular and metabolic pathogenesis
- PMID: 23785136
- PMCID: PMC6618597
- DOI: 10.1523/JNEUROSCI.0822-13.2013
Gangliosides and gangliosidoses: principles of molecular and metabolic pathogenesis
Abstract
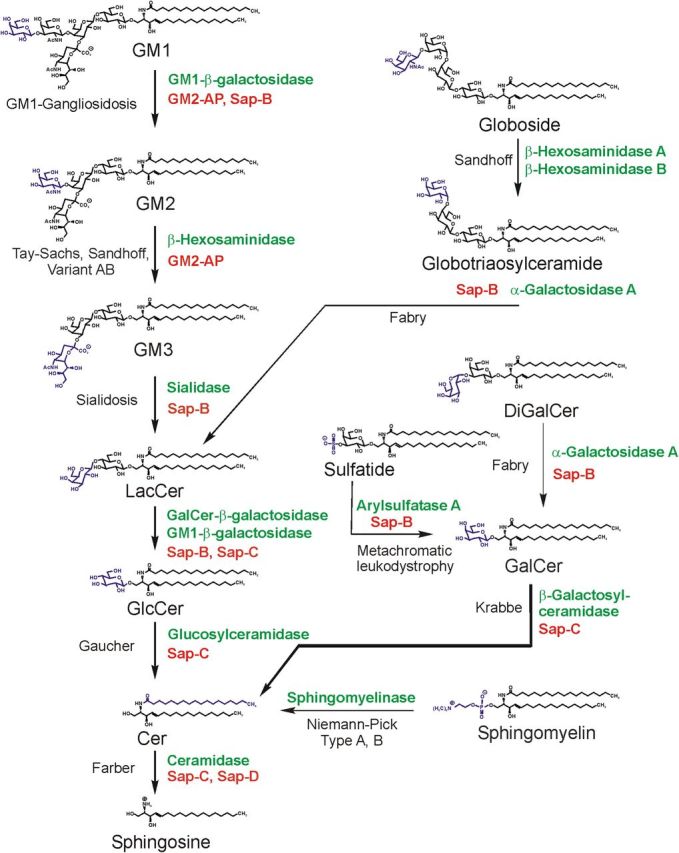
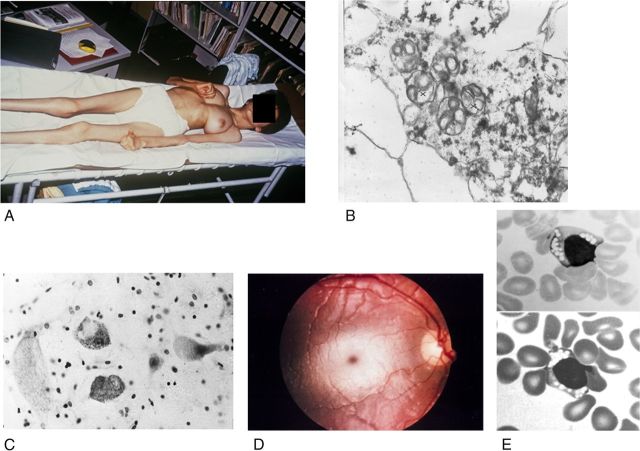
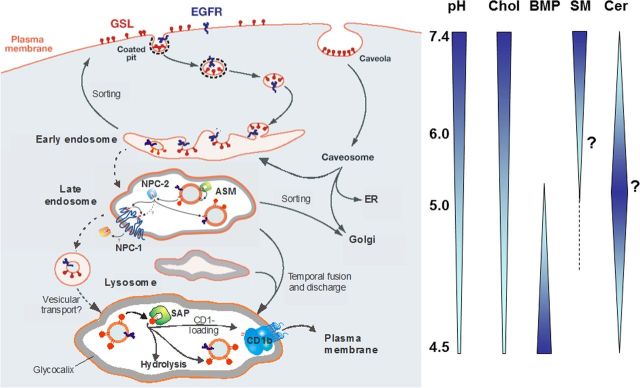
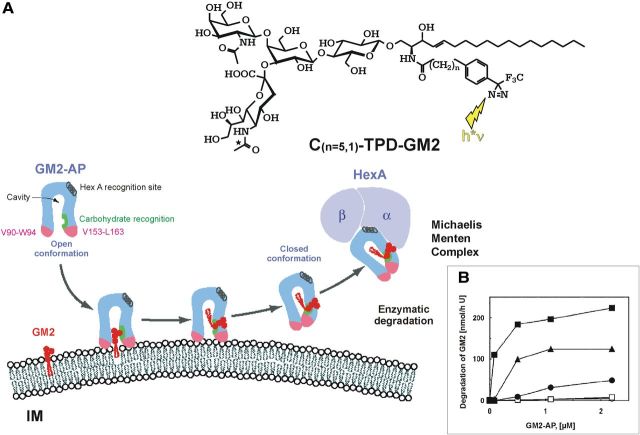
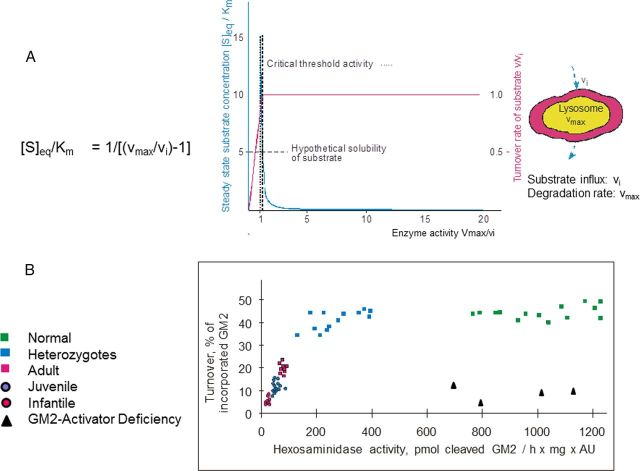
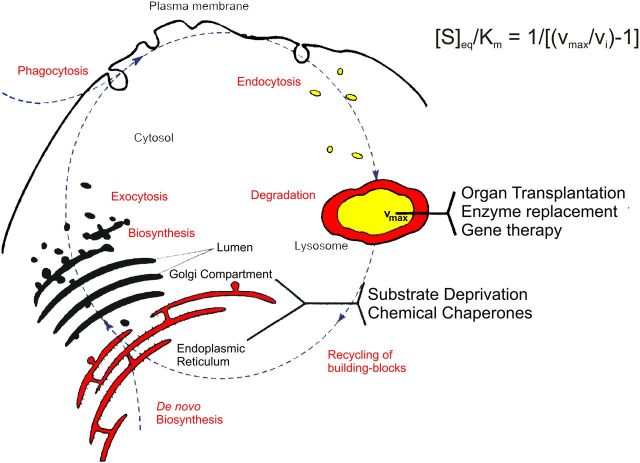
Gangliosides are the main glycolipids of neuronal plasma membranes. Their surface patterns are generated by coordinated processes, involving biosynthetic pathways of the secretory compartments, catabolic steps of the endolysosomal system, and intracellular trafficking. Inherited defects in ganglioside biosynthesis causing fatal neurodegenerative diseases have been described so far almost exclusively in mouse models, whereas inherited defects in ganglioside catabolism causing various clinical forms of GM1- and GM2-gangliosidoses have long been known. For digestion, gangliosides are endocytosed and reach intra-endosomal vesicles. At the level of late endosomes, they are depleted of membrane-stabilizing lipids like cholesterol and enriched with bis(monoacylglycero)phosphate (BMP). Lysosomal catabolism is catalyzed at acidic pH values by cationic sphingolipid activator proteins (SAPs), presenting lipids to their respective hydrolases, electrostatically attracted to the negatively charged surface of the luminal BMP-rich vesicles. Various inherited defects of ganglioside hydrolases, e.g., of β-galactosidase and β-hexosaminidases, and of GM2-activator protein, cause infantile (with tetraparesis, dementia, blindness) and different protracted clinical forms of GM1- and GM2-gangliosidoses. Mutations yielding proteins with small residual catabolic activities in the lysosome give rise to juvenile and adult clinical forms with a wide range of clinical symptomatology. Apart from patients' differences in their genetic background, clinical heterogeneity may be caused by rather diverse substrate specificities and functions of lysosomal hydrolases, multifunctional properties of SAPs, and the strong regulation of ganglioside catabolism by membrane lipids. Currently, there is no treatment available for neuronal ganglioside storage diseases. Therapeutic approaches in mouse models and patients with juvenile forms of gangliosidoses are discussed.
Figures
References
-
- Arfi A, Bourgoin C, Basso L, Emiliani C, Tancini B, Chigorno V, Li YT, Orlacchio A, Poenaru L, Sonnino S, Caillaud C. Bicistronic lentiviral vector corrects beta-hexosaminidase deficiency in transduced and cross-corrected human Sandhoff fibroblasts. Neurobiol Dis. 2005;20:583–593. doi: 10.1016/j.nbd.2005.04.017. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous