

The cystic fibrosis intestine
- PMID: 23788646
- PMCID: PMC3753720
- DOI: 10.1101/cshperspect.a009753
The cystic fibrosis intestine
Abstract
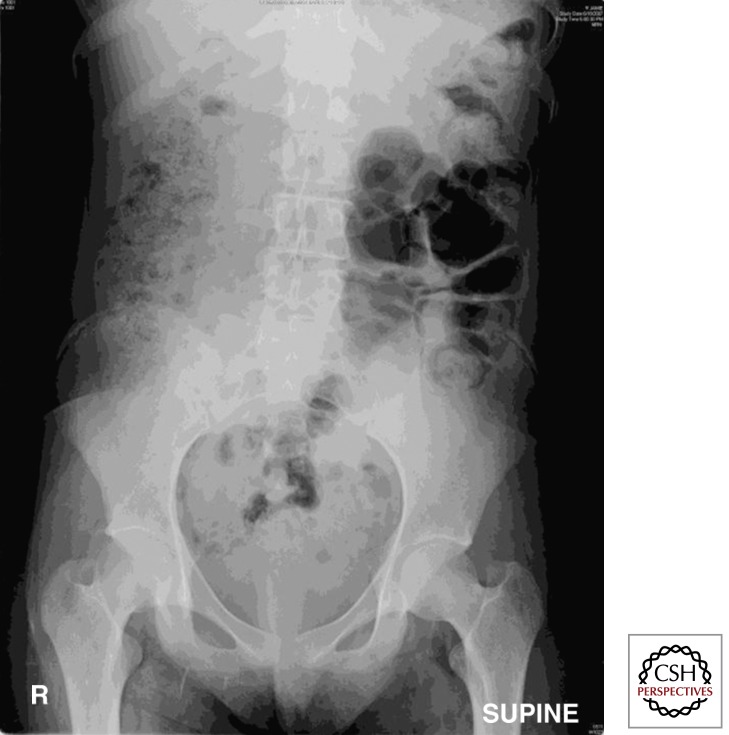
The clinical manifestations of cystic fibrosis (CF) result from dysfunction of the cystic fibrosis transmembrane regulator protein (CFTR). The majority of people with CF have a limited life span as a consequence of CFTR dysfunction in the respiratory tract. However, CFTR dysfunction in the gastrointestinal (GI) tract occurs earlier in ontogeny and is present in all patients, regardless of genotype. The same pathophysiologic triad of obstruction, infection, and inflammation that causes disease in the airways also causes disease in the intestines. This article describes the effects of CFTR dysfunction on the intestinal tissues and the intraluminal environment. Mouse models of CF have greatly advanced our understanding of the GI manifestations of CF, which can be directly applied to understanding CF disease in humans.
Figures

References
-
- Baker SS, Borowitz D, Duffy L, Fitzpatrick L, Gyamfi J, Baker RD 2005. Pancreatic enzyme therapy and clinical outcomes in patients with cystic fibrosis. J Pediatr 146: 189–193 - PubMed
-
- Barraclough M, Taylor CJ 1996. Twenty-four hour ambulatory gastric and duodenal pH profiles in cystic fibrosis: Effect of duodenal hyperacidity on pancreatic enzyme function and fat absorption. J Pediatr Gastroenterol Nutr 23: 45–50 - PubMed
-
- Beharry S, Ackerley C, Corey M, Kent G, Heng YM, Christensen H, Luk C, Yantiss RK, Nasser IA, Zaman M, et al. 2007. Long-term docosahexaenoic acid therapy in a congenic murine model of cystic fibrosis. Am J Physiol Gastrointest Liver Physiol 292: G839–G848 - PubMed
-
- Bhardwaj S, Canlas K, Kahi C, Temkit M, Molleston J, Ober M, Howenstine M, Kwo PY 2009. Hepatobiliary abnormalities and disease in cystic fibrosis: Epidemiology and outcomes through adulthood. J Clin Gastroenterol 43: 858–864 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical