

Mitochondrial oxidative stress corrupts coronary collateral growth by activating adenosine monophosphate activated kinase-α signaling
- PMID: 23788766
- PMCID: PMC4402936
- DOI: 10.1161/ATVBAHA.113.301591
Mitochondrial oxidative stress corrupts coronary collateral growth by activating adenosine monophosphate activated kinase-α signaling
Abstract
Objective: Our goal was to determine the mechanism by which mitochondrial oxidative stress impairs collateral growth in the heart.
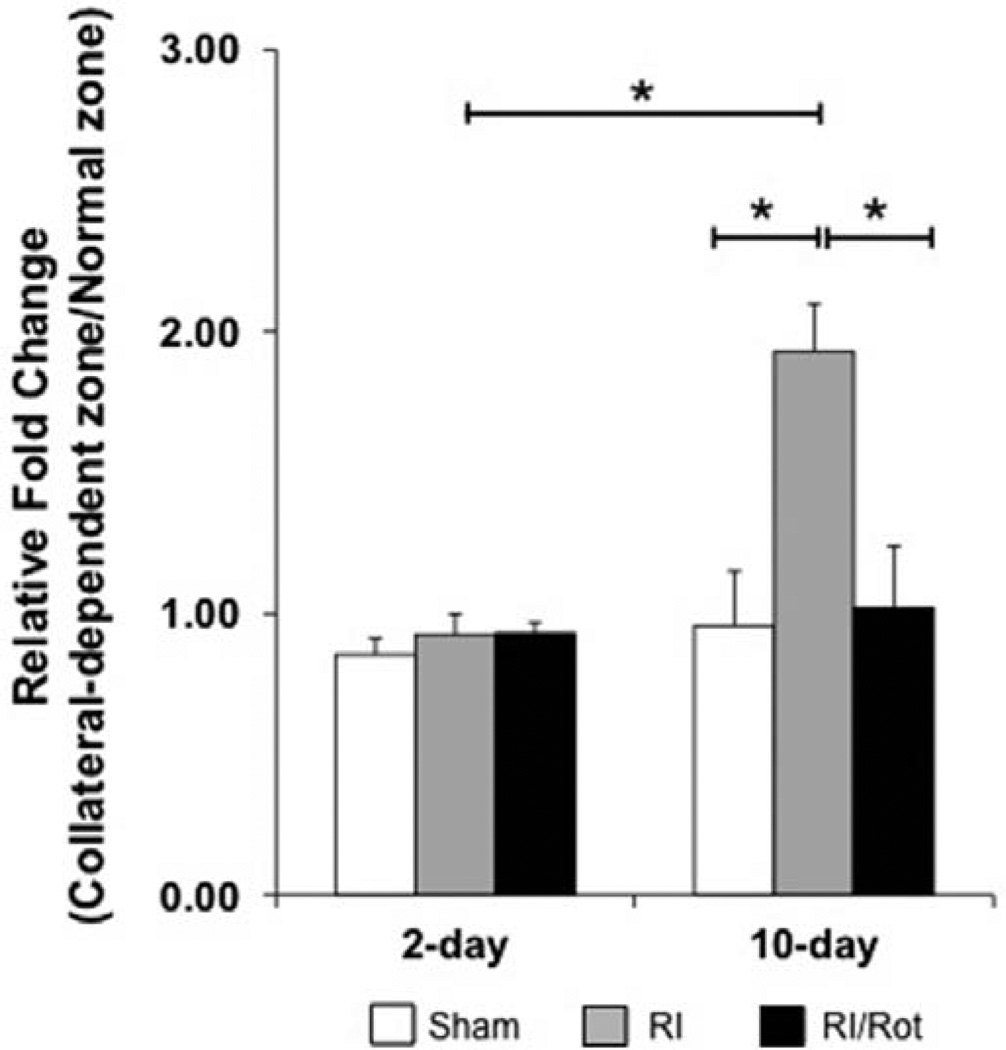
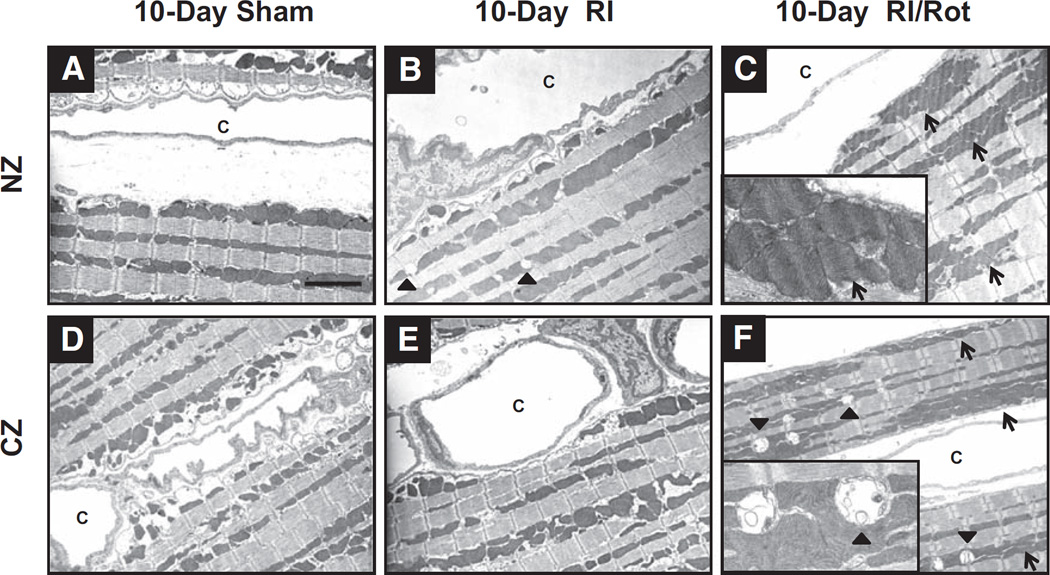
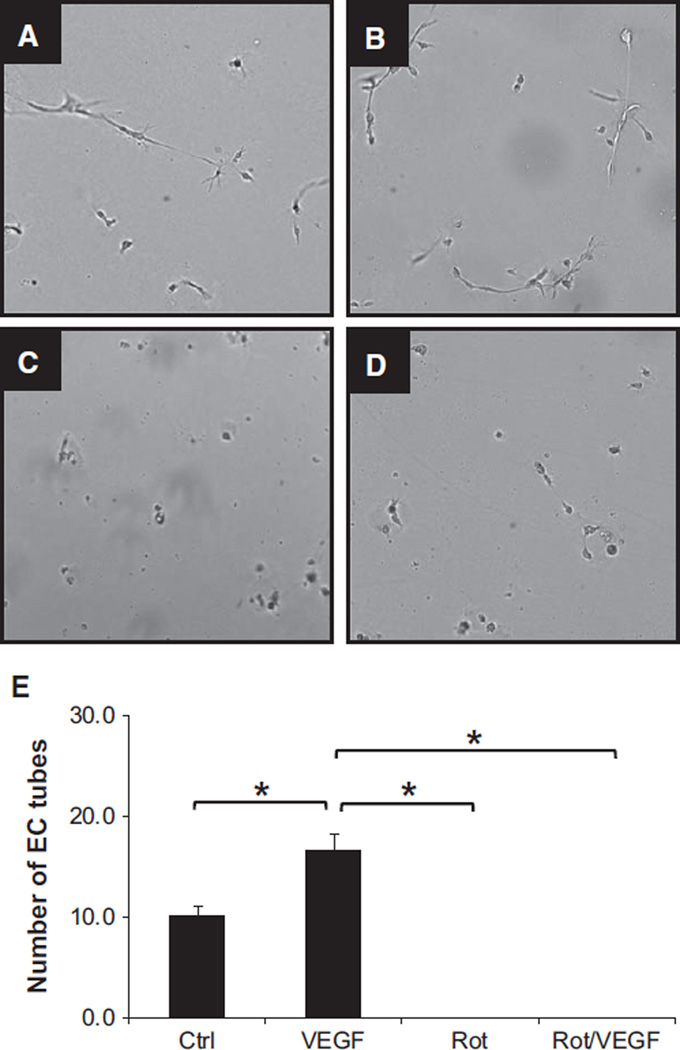
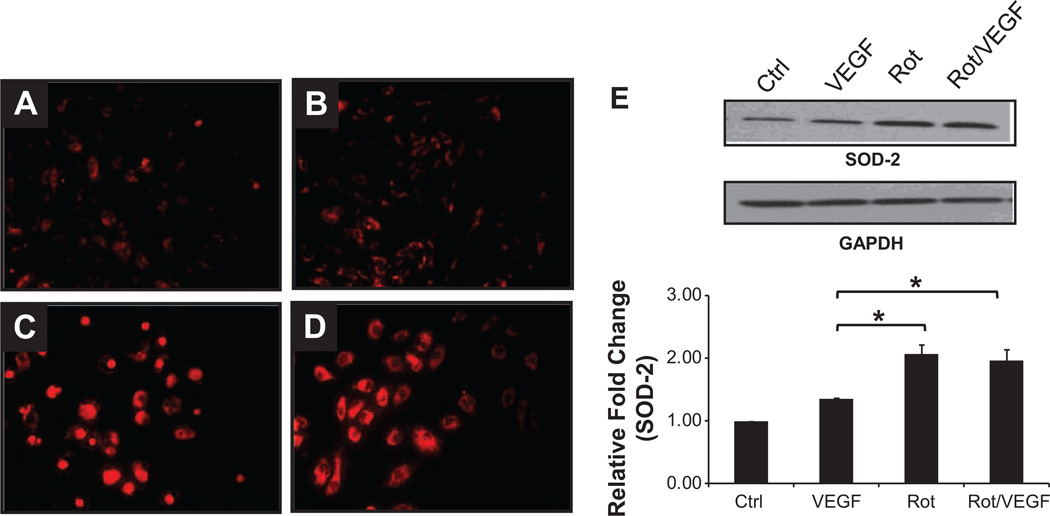
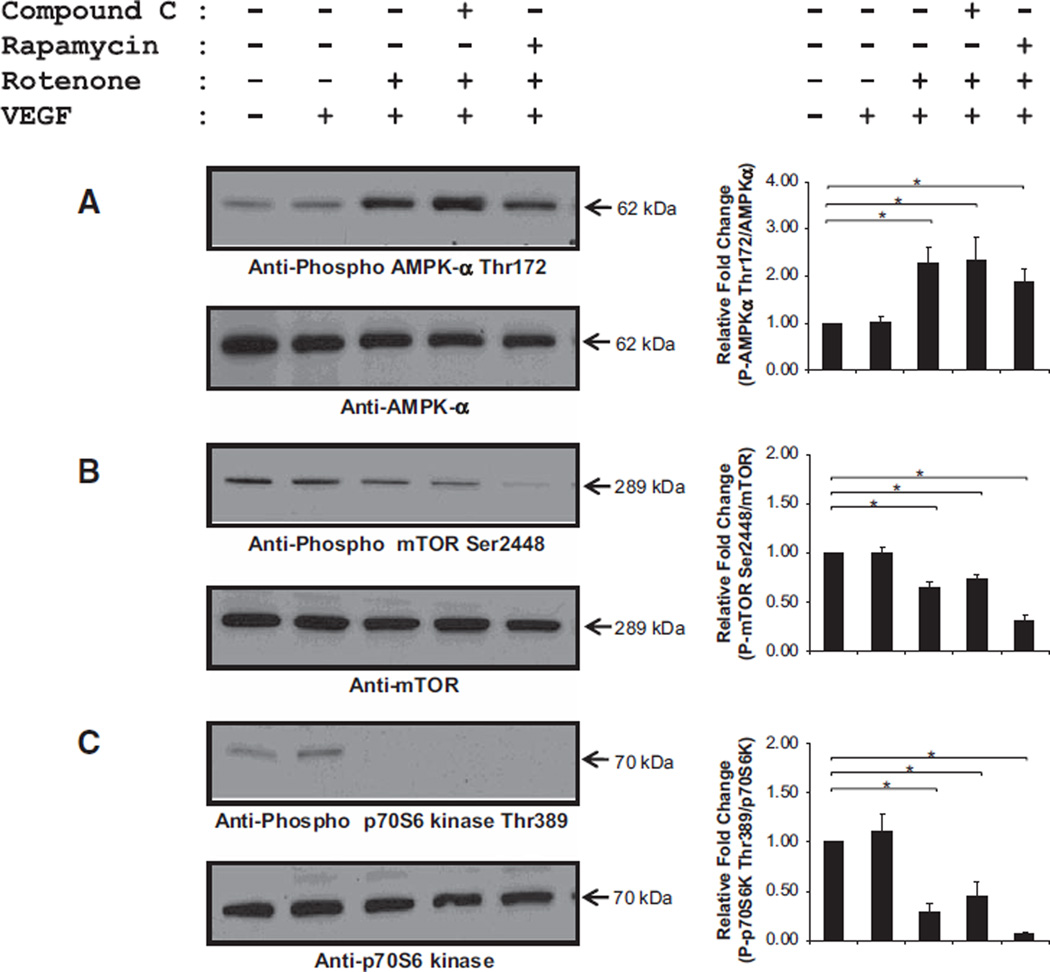
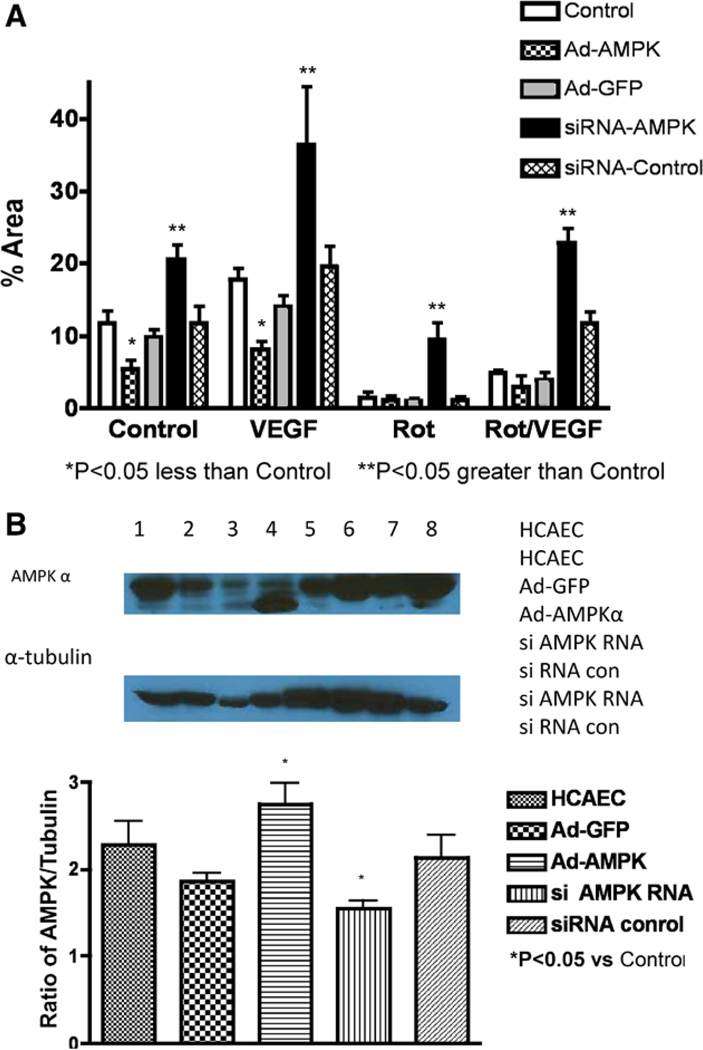
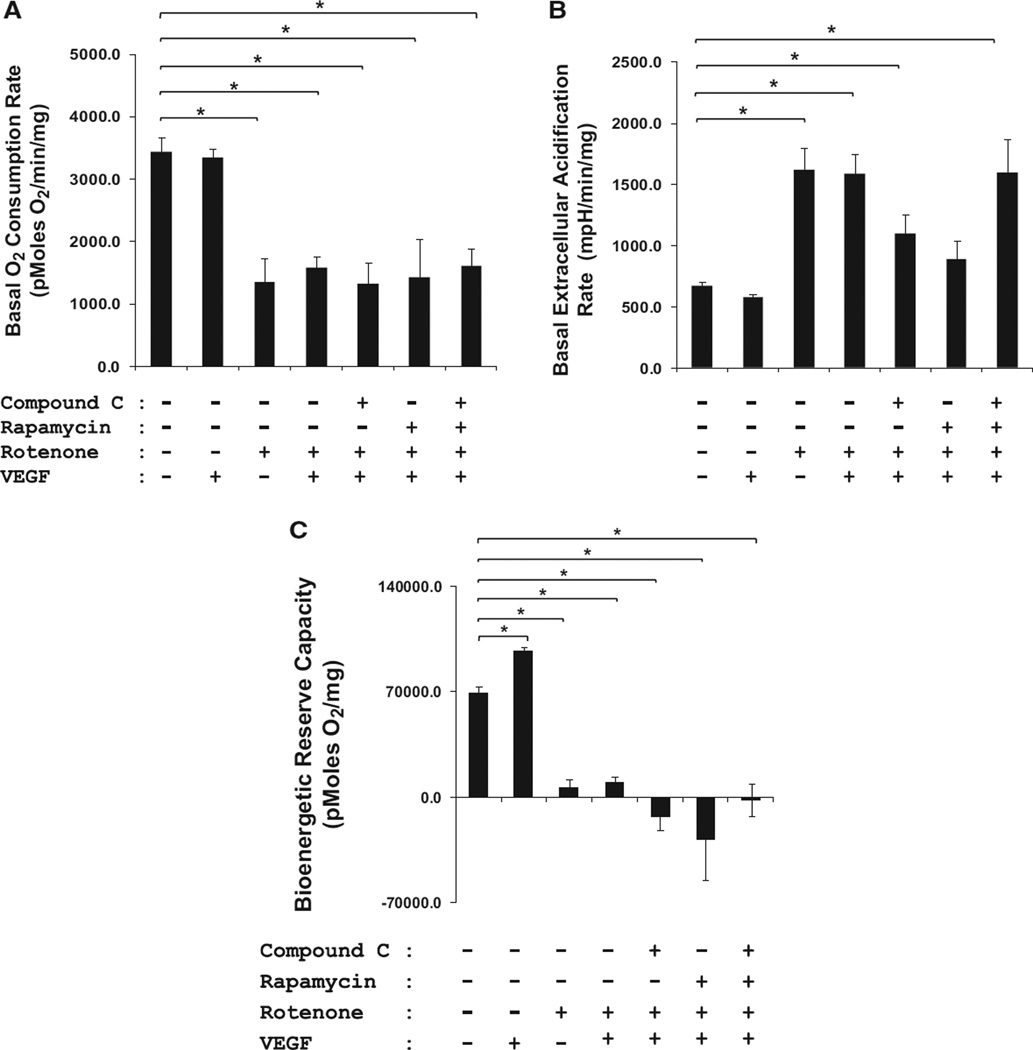
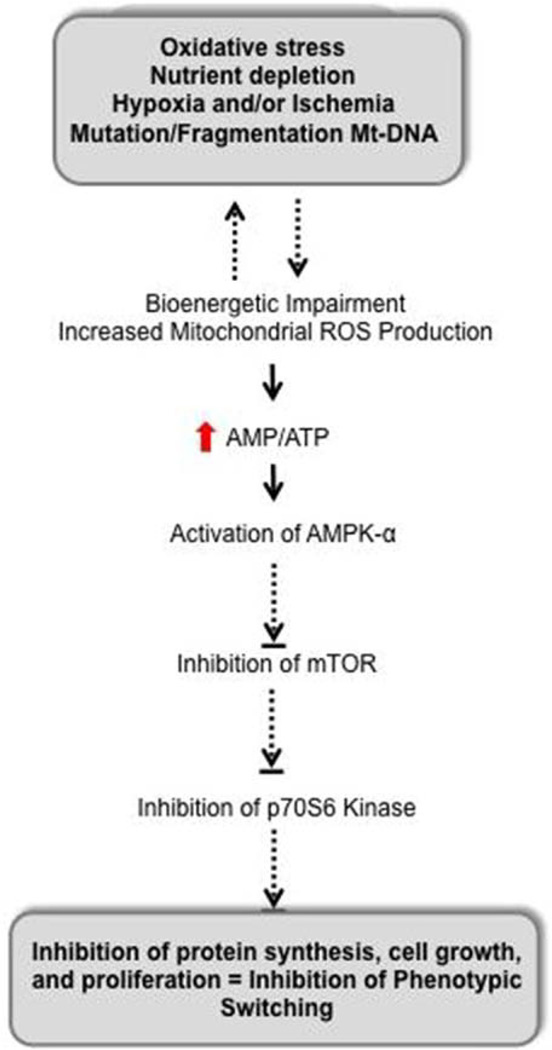
Approach and results: Rats were treated with rotenone (mitochondrial complex I inhibitor that increases reactive oxygen species production) or sham-treated with vehicle and subjected to repetitive ischemia protocol for 10 days to induce coronary collateral growth. In control rats, repetitive ischemia increased flow to the collateral-dependent zone; however, rotenone treatment prevented this increase suggesting that mitochondrial oxidative stress compromises coronary collateral growth. In addition, rotenone also attenuated mitochondrial complex I activity and led to excessive mitochondrial aggregation. To further understand the mechanistic pathway(s) involved, human coronary artery endothelial cells were treated with 50 ng/mL vascular endothelial growth factor, 1 µmol/L rotenone, and rotenone/vascular endothelial growth factor for 48 hours. Vascular endothelial growth factor induced robust tube formation; however, rotenone completely inhibited this effect (P<0.05 rotenone versus vascular endothelial growth factor treatment). Inhibition of tube formation by rotenone was also associated with significant increase in mitochondrial superoxide generation. Immunoblot analyses of human coronary artery endothelial cells with rotenone treatment showed significant activation of adenosine monophosphate activated kinase (AMPK)-α and inhibition of mammalian target of rapamycin and p70 ribosomal S6 kinase. Activation of AMPK-α suggested impairments in energy production, which was reflected by decrease in O2 consumption and bioenergetic reserve capacity of cultured cells. Knockdown of AMPK-α (siRNA) also preserved tube formation during rotenone, suggesting the negative effects were mediated by the activation of AMPK-α. Conversely, expression of a constitutively active AMPK-α blocked tube formation.
Conclusions: We conclude that activation of AMPK-α during mitochondrial oxidative stress inhibits mammalian target of rapamycin signaling, which impairs phenotypic switching necessary for the growth of blood vessels.
Keywords: collateral circulation; coronary circulation; mitochondria; reactive oxygen species.
Figures
References
-
- Bugger H, Abel ED. Molecular mechanisms for myocardial mitochondrial dysfunction in the metabolic syndrome. Clin Sci (Lond) 2008;114:195–210. - PubMed
-
- Magliano DJ, Shaw JE, Zimmet PZ. How to best define the metabolic syndrome. Ann Med. 2006;38:34–41. - PubMed
-
- Zimmet P, Magliano D, Matsuzawa Y, Alberti G, Shaw J. The metabolic syndrome: a global public health problem and a new definition. J Atheroscler Thromb. 2005;12:295–300. - PubMed
-
- Ford ES, Giles WH, Dietz WH. Prevalence of the metabolic syndrome among US adults: findings from the third National Health and Nutrition Examination Survey. JAMA. 2002;287:356–359. - PubMed
-
- Abaci A, Oğuzhan A, Kahraman S, Eryol NK, Unal S, Arinç H, Ergin A. Effect of diabetes mellitus on formation of coronary collateral vessels. Circulation. 1999;99:2239–2242. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
