

Massive genomic variation and strong selection in Arabidopsis thaliana lines from Sweden
- PMID: 23793030
- PMCID: PMC3755268
- DOI: 10.1038/ng.2678
Massive genomic variation and strong selection in Arabidopsis thaliana lines from Sweden
Abstract
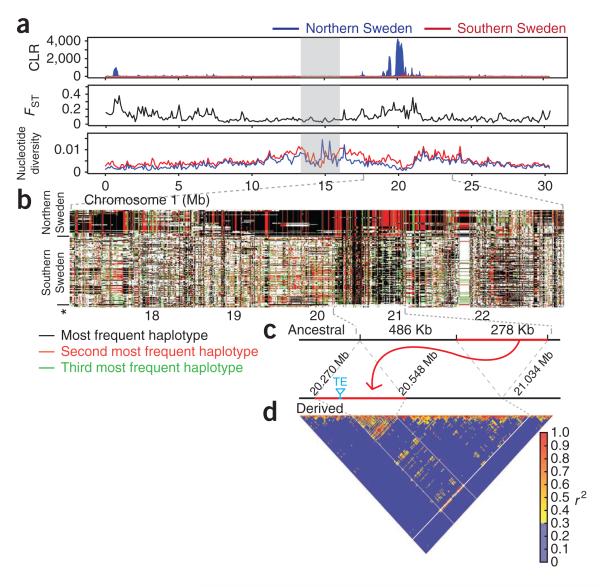
Despite advances in sequencing, the goal of obtaining a comprehensive view of genetic variation in populations is still far from reached. We sequenced 180 lines of A. thaliana from Sweden to obtain as complete a picture as possible of variation in a single region. Whereas simple polymorphisms in the unique portion of the genome are readily identified, other polymorphisms are not. The massive variation in genome size identified by flow cytometry seems largely to be due to 45S rDNA copy number variation, with lines from northern Sweden having particularly large numbers of copies. Strong selection is evident in the form of long-range linkage disequilibrium (LD), as well as in LD between nearby compensatory mutations. Many footprints of selective sweeps were found in lines from northern Sweden, and a massive global sweep was shown to have involved a 700-kb transposition.
Figures
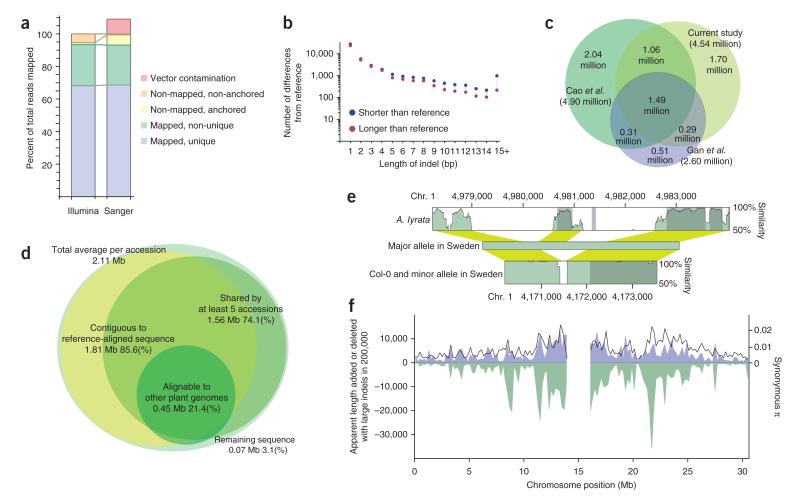
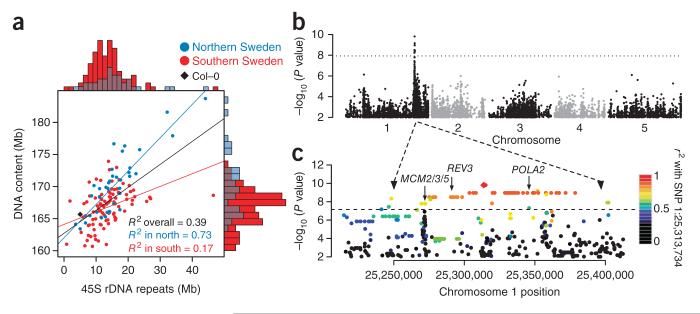
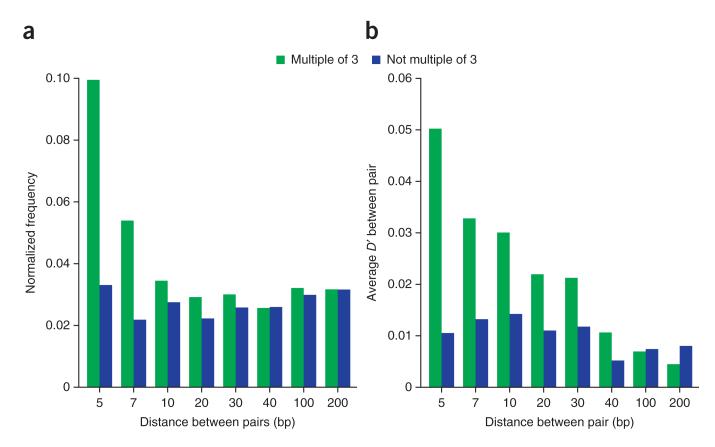
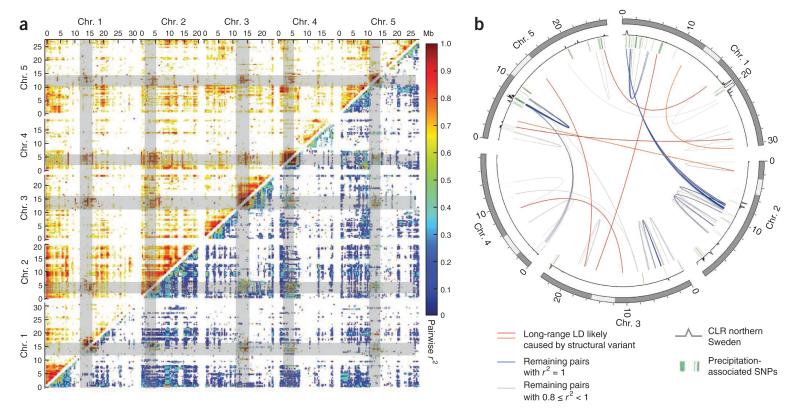
References
-
- Fournier-Level A, et al. A map of local adaptation in Arabidopsis thaliana. Science. 2011;334:86–89. - PubMed
-
- Hancock AM, et al. Adaptation to climate across the Arabidopsis thaliana genome. Science. 2011;334:83–86. - PubMed
-
- Koornneef M, Alonso-Blanco C, Vreugdenhil D. Naturally occurring genetic variation in Arabidopsis thaliana. Annu. Rev. Plant Biol. 2004;55:141–172. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
