

GFPT1-myasthenia: clinical, structural, and electrophysiologic heterogeneity
- PMID: 23794683
- PMCID: PMC3772836
- DOI: 10.1212/WNL.0b013e31829c5e9c
GFPT1-myasthenia: clinical, structural, and electrophysiologic heterogeneity
Abstract
Objective: To identify patients with GFPT1-related limb-girdle myasthenia and analyze phenotypic consequences of the mutations.
Methods: We performed genetic analysis, histochemical, immunoblot, and ultrastructural studies and in vitro electrophysiologic analysis of neuromuscular transmission.
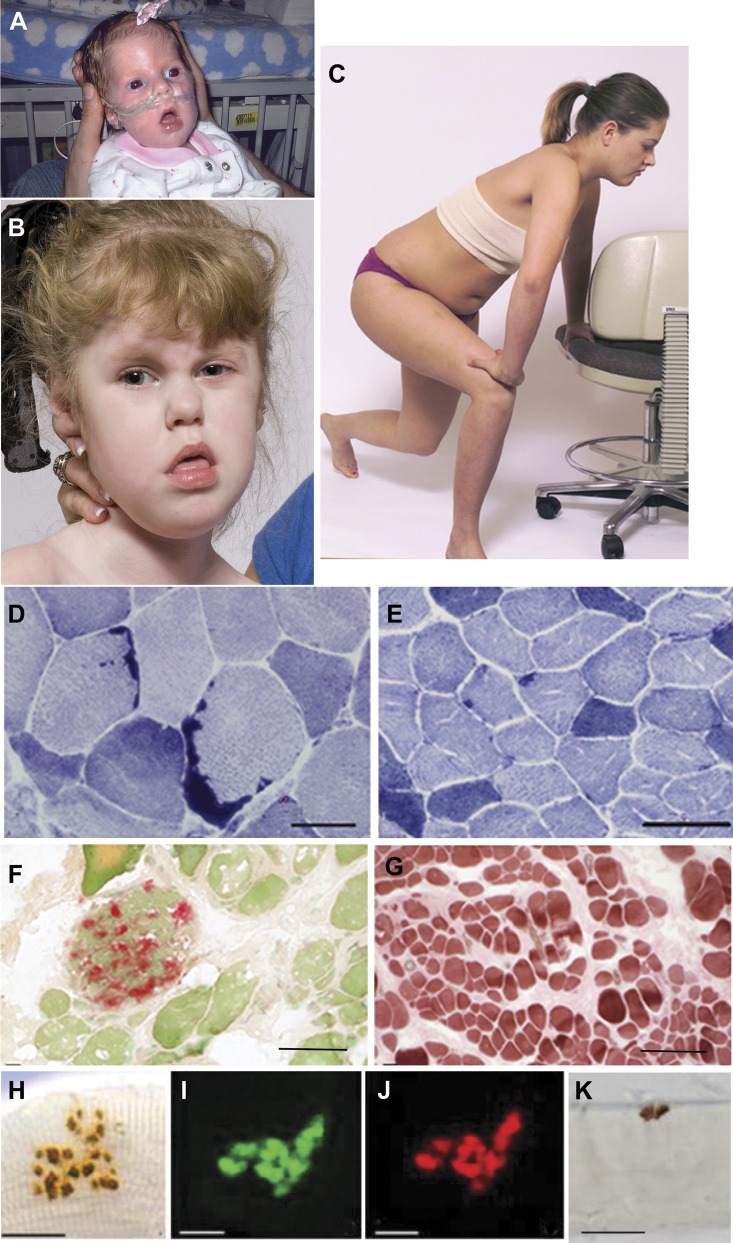
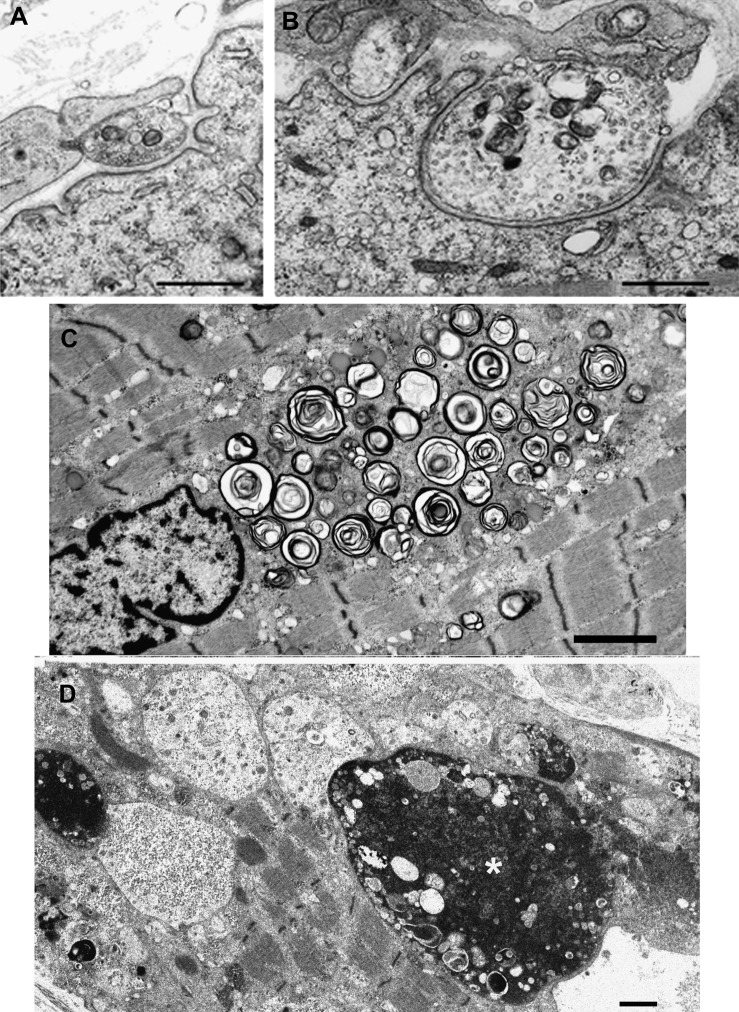
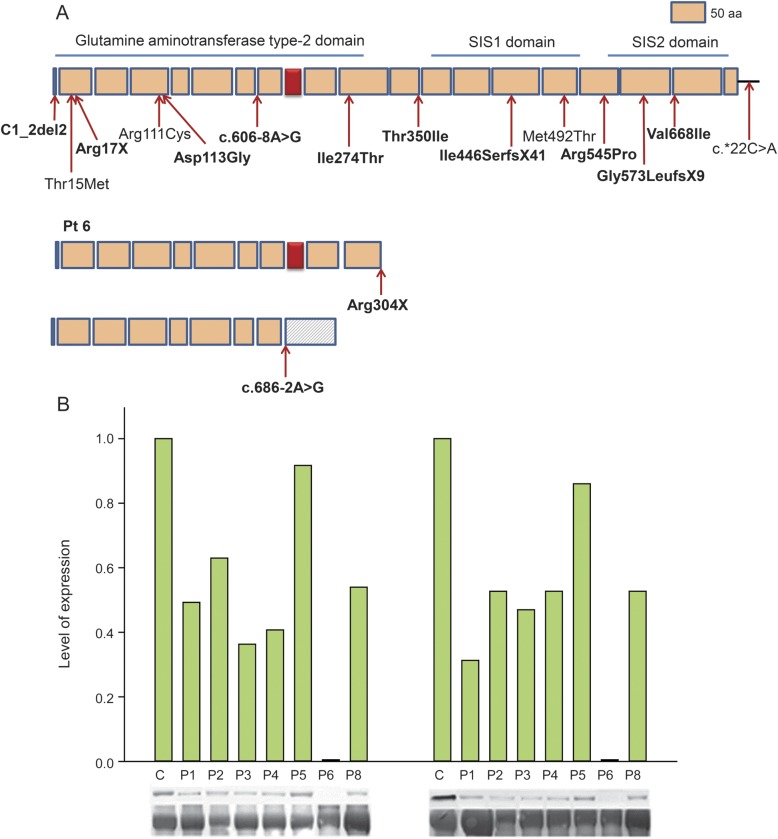
Results: We identified 16 recessive mutations in GFPT1 in 11 patients, of which 12 are novel. Ten patients had slowly progressive limb-girdle weakness responsive to cholinergic agonists with onset between infancy and age 19 years. One patient (no. 6) harbored a nonsense mutation and a second mutation that disrupts the muscle-specific GFPT1 exon. This patient never moved in utero, was apneic and arthrogrypotic at birth, and was bedfast, tube-fed, and barely responded to therapy at age 6 years. Histochemical studies in 9 of 11 patients showed tubular aggregates in 6 and rimmed vacuoles in 3. Microelectrode studies of intercostal muscle endplates in 5 patients indicated reduced synaptic response to acetylcholine in 3 and severely reduced quantal release in patient 6. Endplate acetylcholine receptor content was moderately reduced in only one patient. The synaptic contacts were small and single or grape-like, and quantitative electron microscopy revealed hypoplastic endplate regions. Numerous muscle fibers of patient 6 contained myriad dilated and degenerate vesicular profiles, autophagic vacuoles, and bizarre apoptotic nuclei. Glycoprotein expression in muscle was absent in patient 6 and reduced in 5 others.
Conclusions: GFPT1-myasthenia is more heterogeneous than previously reported. Different parameters of neuromuscular transmission are variably affected. When disruption of muscle-specific isoform determines the phenotype, this has devastating clinical, pathologic, and biochemical consequences.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous