

Computational methods for constructing protein structure models from 3D electron microscopy maps
- PMID: 23796504
- PMCID: PMC3795986
- DOI: 10.1016/j.jsb.2013.06.008
Computational methods for constructing protein structure models from 3D electron microscopy maps
Abstract
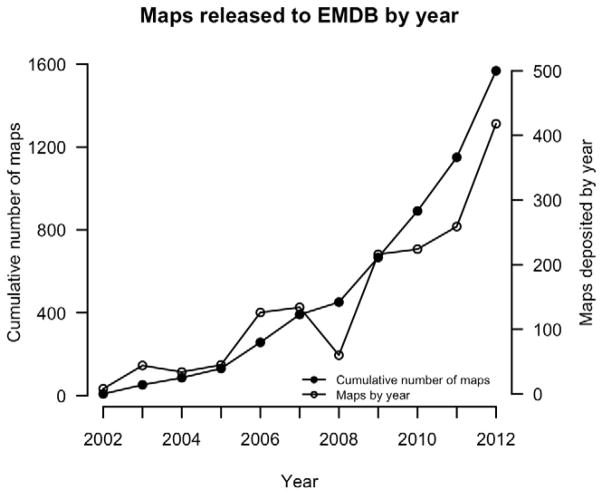
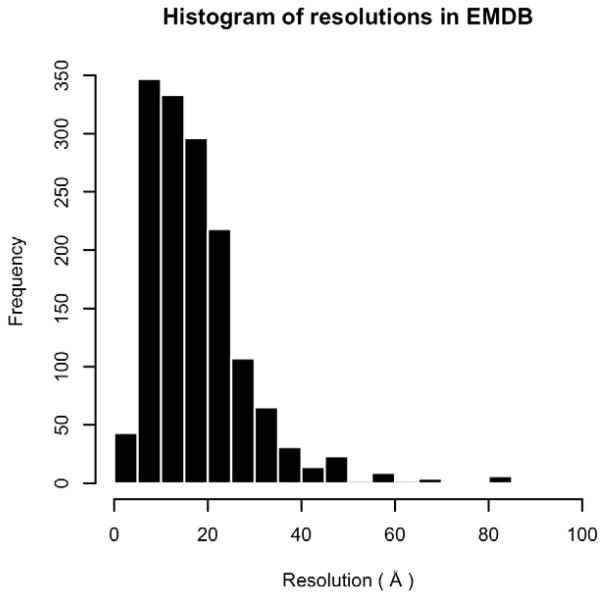
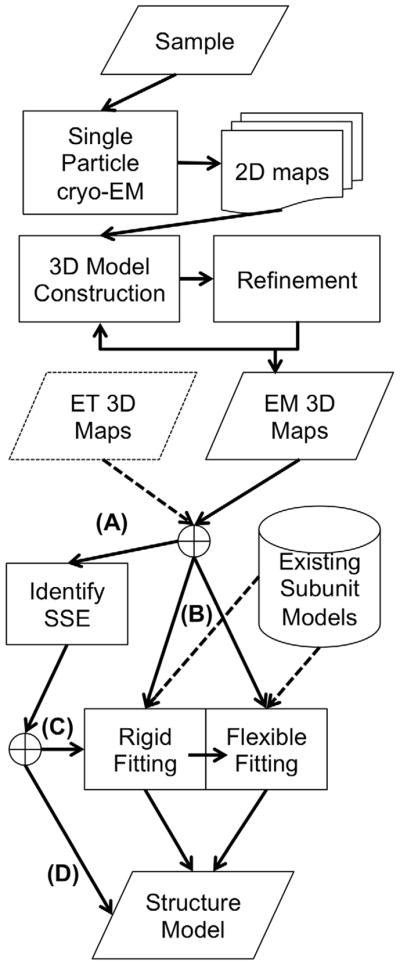
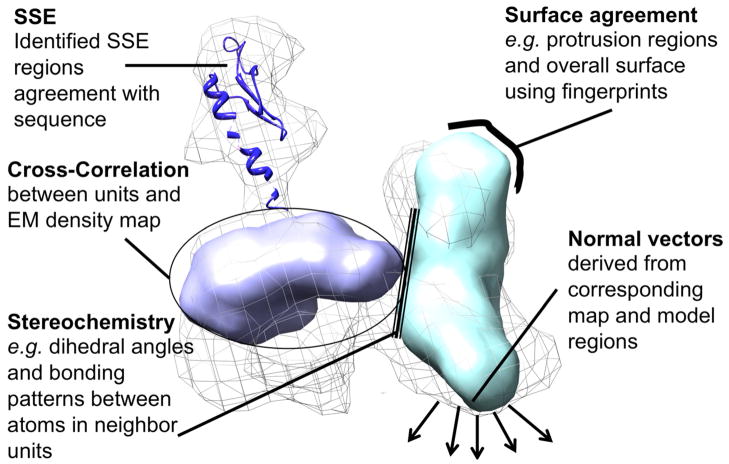
Protein structure determination by cryo-electron microscopy (EM) has made significant progress in the past decades. Resolutions of EM maps have been improving as evidenced by recently reported structures that are solved at high resolutions close to 3Å. Computational methods play a key role in interpreting EM data. Among many computational procedures applied to an EM map to obtain protein structure information, in this article we focus on reviewing computational methods that model protein three-dimensional (3D) structures from a 3D EM density map that is constructed from two-dimensional (2D) maps. The computational methods we discuss range from de novo methods, which identify structural elements in an EM map, to structure fitting methods, where known high resolution structures are fit into a low-resolution EM map. A list of available computational tools is also provided.
Keywords: 3D Zernike descriptor; 3DZD; CATH; Class, Architecture, Topology, Homologous superfamily. Acronym for the CATH protein structure database; Computational algorithm; DEN; Deformable Elastic Network; EM; EMDB; ENM; Elastic Network Model; Electron Microscopy Data Bank; Electron density map; Electron microscopy; MC; MD; MDFF; Macromolecular structure modeling; Monte Carlo; NMA; NMFF; NMR; Normal Mode Flexible Fitting; Nuclear Magnetic Resonance; PDB; Protein Data Bank; RMSD; Root Mean Square Deviation; SVM; Structure fitting; electron microscopy; molecular dynamics; molecular dynamics flexible fitting; normal mode analysis; support vector machine.
Copyright © 2013 Elsevier Inc. All rights reserved.
Figures
References
-
- Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006;22:195–201. - PubMed
-
- Bahar I, Atilgan AR, Erman B. Direct evaluation of thermal fluctuations in proteins using a single-parameter harmonic potential. Fold Des. 1997;2:173–81. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
