

The respiratory neuromuscular system in Pompe disease
- PMID: 23797185
- PMCID: PMC4083814
- DOI: 10.1016/j.resp.2013.06.007
The respiratory neuromuscular system in Pompe disease
Abstract
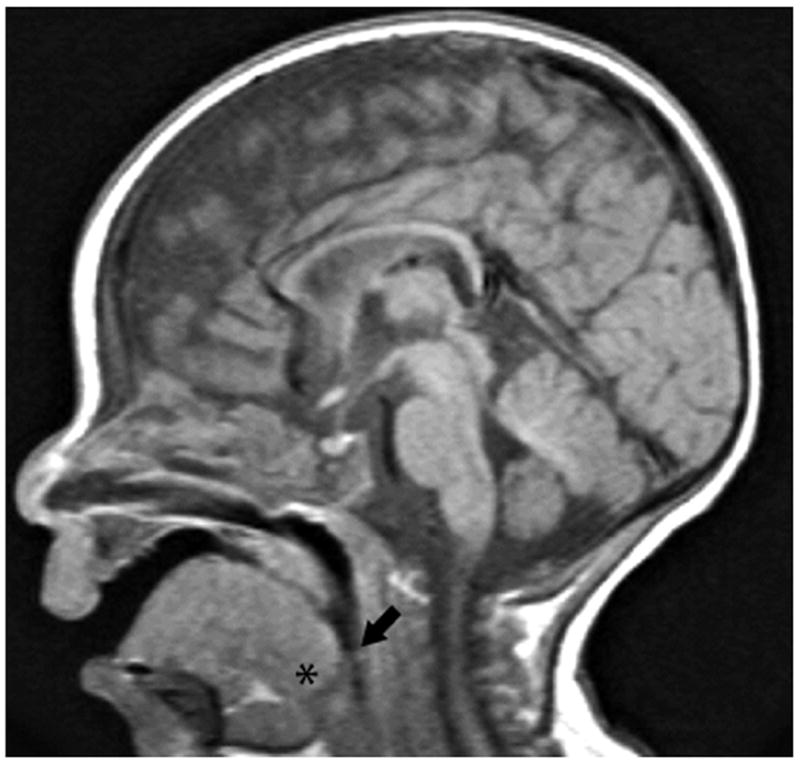
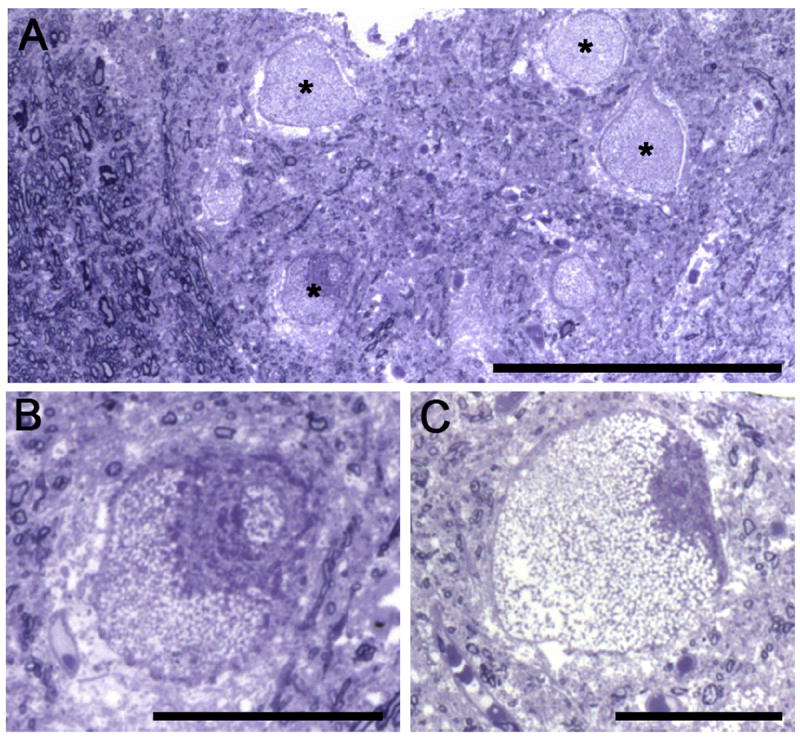
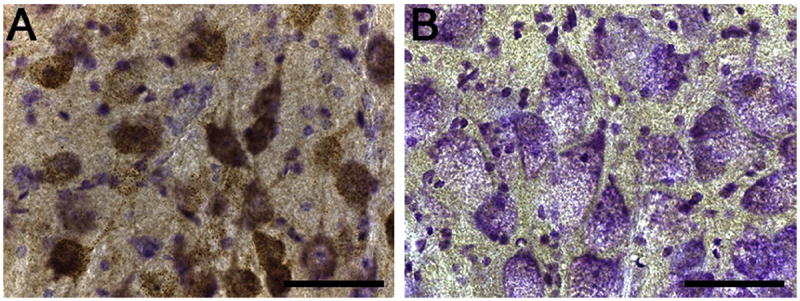
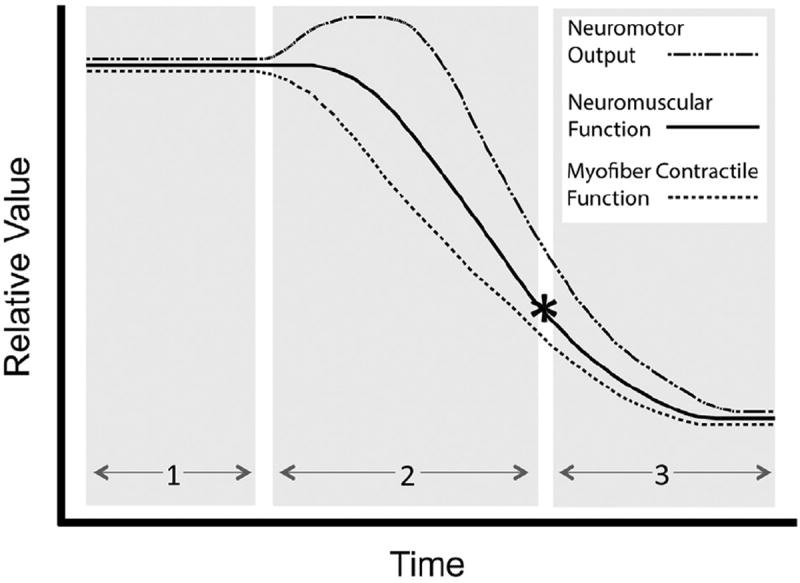
Pompe disease is due to mutations in the gene encoding the lysosomal enzyme acid α-glucosidase (GAA). Absence of functional GAA typically results in cardiorespiratory failure in the first year; reduced GAA activity is associated with progressive respiratory failure later in life. While skeletal muscle pathology contributes to respiratory insufficiency in Pompe disease, emerging evidence indicates that respiratory neuron dysfunction is also a significant part of dysfunction in motor units. Animal models show profound glycogen accumulation in spinal and medullary respiratory neurons and altered neural activity. Tissues from Pompe patients show central nervous system glycogen accumulation and motoneuron pathology. A neural mechanism raises considerations about the current clinical approach of enzyme replacement since the recombinant protein does not cross the blood-brain-barrier. Indeed, clinical data suggest that enzyme replacement therapy delays symptom progression, but many patients eventually require ventilatory assistance, especially during sleep. We propose that treatments which restore GAA activity to respiratory muscles, neurons and networks will be required to fully correct ventilatory insufficiency in Pompe disease.
Keywords: Motoneurons; Pathology; Plasticity; Pompe; Respiratory; Therapy.
Copyright © 2013. Published by Elsevier B.V.
Figures
References
-
- Amalfitano A, Bengur AR, Morse RP, Majure JM, Case LE, Veerling DL, Mackey J, Kishnani P, Smith W, McVie-Wylie A, Sullivan JA, Hoganson GE, Phillips JA, 3rd, Schaefer GB, Charrow J, Ware RE, Bossen EH, Chen YT. Recombinant human acid alpha-glucosidase enzyme therapy for infantile glycogen storage disease type II: results of a phase I/II clinical trial. Genetics in Medicine: Official Journal of the American College of Medical Genetics. 2001;3:132–138. - PubMed
-
- Bailey EF, Fregosi RF. Coordination of intrinsic and extrinsic tongue muscles during spontaneous breathing in the rat. Journal of Applied Physiology. 2004;96:440–449. - PubMed
-
- Baudhuin P, Hers HG, Loeb H. An electron microscopic and biochemical study of type Ii glycogenosis. Laboratory Investigation. 1964;13:1139–1152. - PubMed
-
- Beck M. Therapy for lysosomal storage disorders. IUBMB Life. 2010;62:33–40. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
