

The role of JAK-3 in regulating TLR-mediated inflammatory cytokine production in innate immune cells
- PMID: 23797672
- PMCID: PMC3720823
- DOI: 10.4049/jimmunol.1203084
The role of JAK-3 in regulating TLR-mediated inflammatory cytokine production in innate immune cells
Abstract
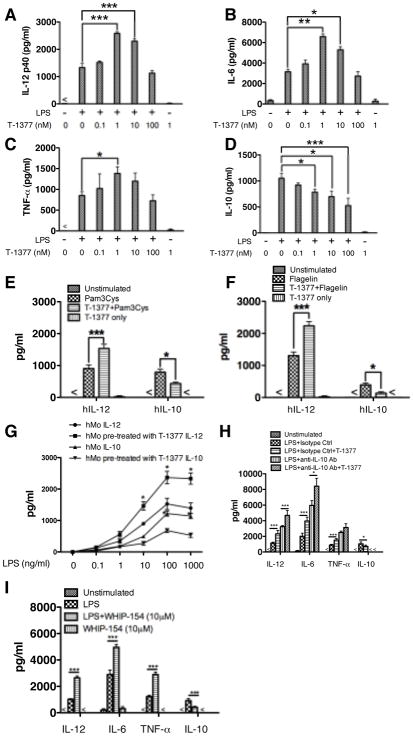
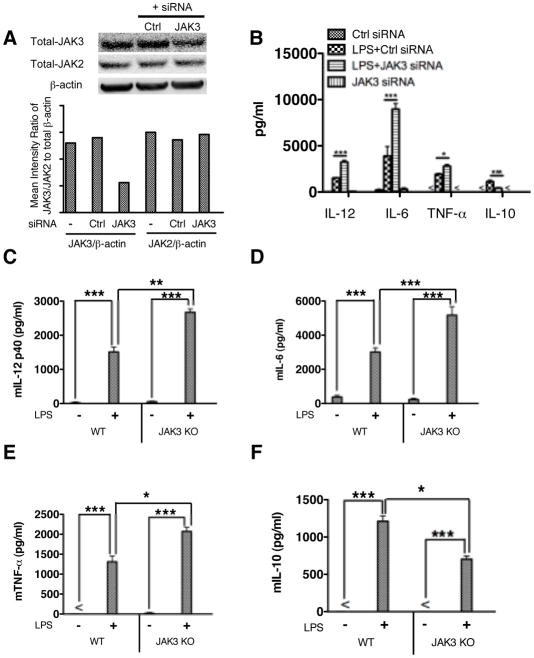
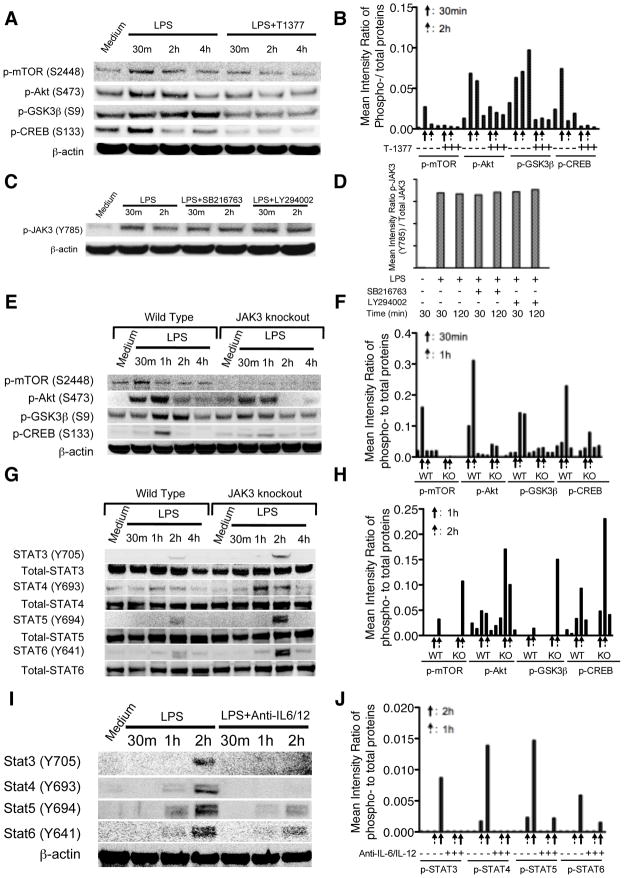
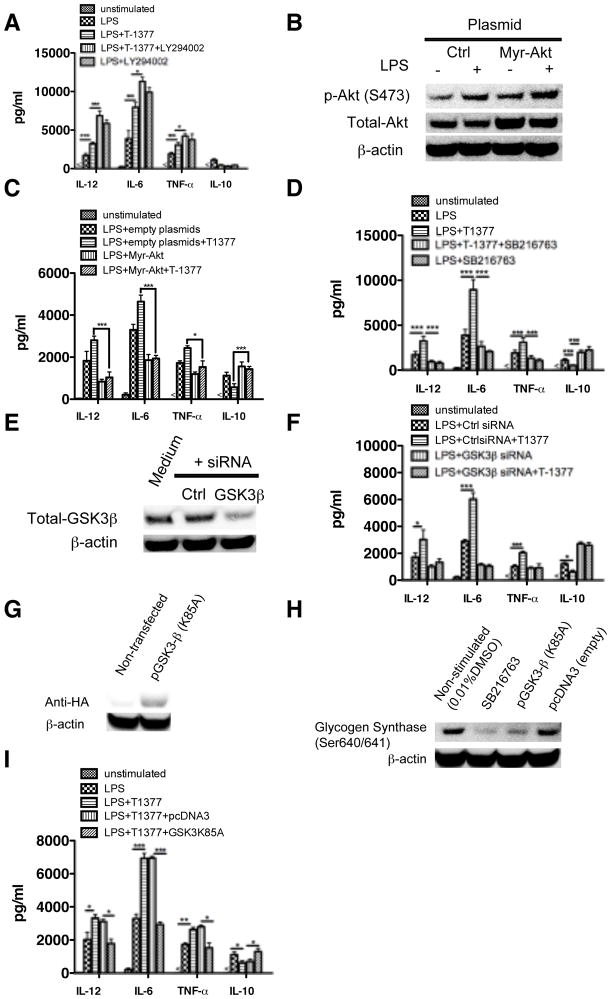
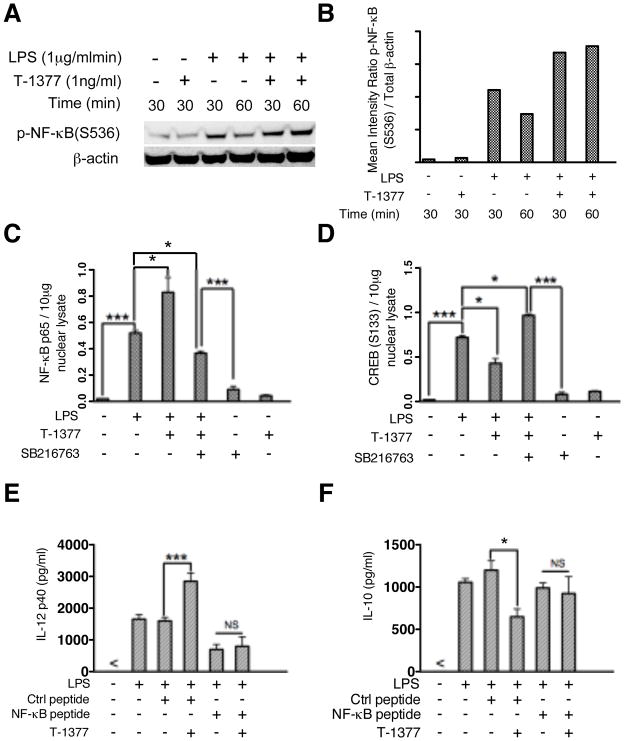
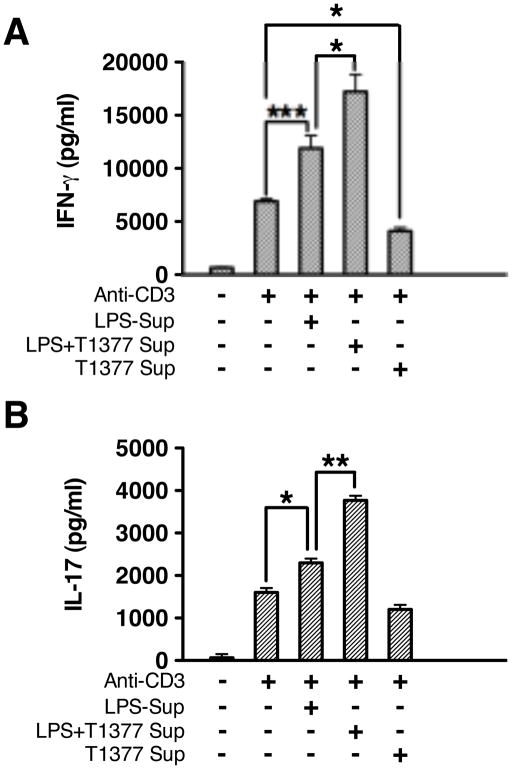
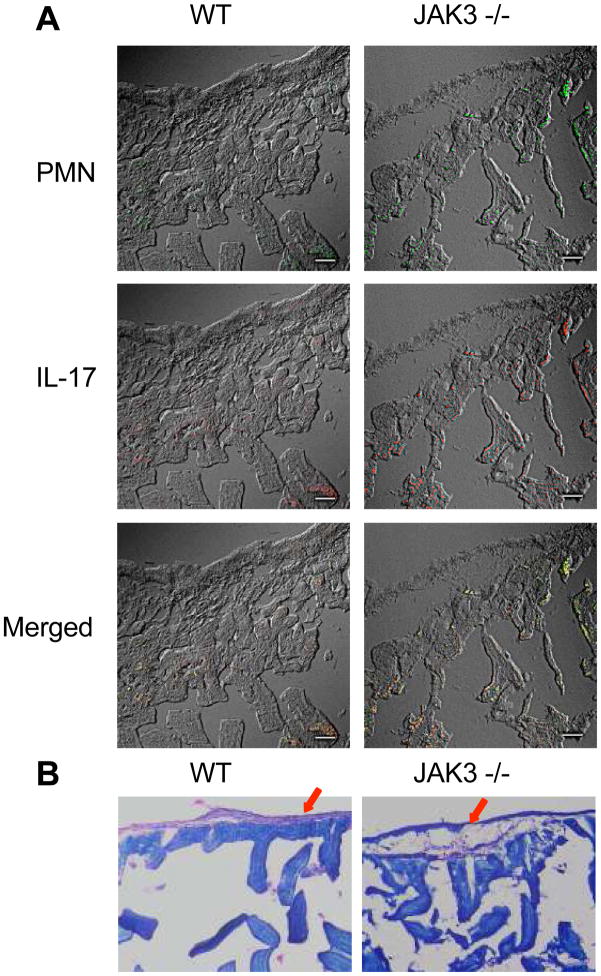
The role of JAK-3 in TLR-mediated innate immune responses is poorly understood, although the suppressive function of JAK3 inhibition in adaptive immune response has been well studied. In this study, we found that JAK3 inhibition enhanced TLR-mediated immune responses by differentially regulating pro- and anti- inflammatory cytokine production in innate immune cells. Specifically, JAK3 inhibition by pharmacological inhibitors or specific small interfering RNA or JAK3 gene knockout resulted in an increase in TLR-mediated production of proinflammatory cytokines while concurrently decreasing the production of IL-10. Inhibition of JAK3 suppressed phosphorylation of PI3K downstream effectors including Akt, mammalian target of rapamycin complex 1, glycogen synthase kinase 3β (GSK3β), and CREB. Constitutive activation of Akt or inhibition of GSK3β abrogated the capability of JAK3 inhibition to enhance proinflammatory cytokines and suppress IL-10 production. In contrast, inhibition of PI3K enhanced this regulatory ability of JAK3 in LPS-stimulated monocytes. At the transcriptional level, JAK3 knockout lead to the increased phosphorylation of STATs that could be attenuated by neutralization of de novo inflammatory cytokines. JAK3 inhibition exhibited a GSK3 activity-dependent ability to enhance phosphorylation levels and DNA binding of NF-κB p65. Moreover, JAK3 inhibition correlated with an increased CD4(+) T cell response. Additionally, higher neutrophil infiltration, IL-17 expression, and intestinal epithelium erosion were observed in JAK3 knockout mice. These findings demonstrate the negative regulatory function of JAK3 and elucidate the signaling pathway by which JAK3 differentially regulates TLR-mediated inflammatory cytokine production in innate immune cells.
Conflict of interest statement
Conflict-of-Interest Disclosure
The authors declare no competing financial interests
Figures
References
-
- O’Neill LA. ‘Fine tuning’ TLR signaling. Nat Immunol. 2008;9:459–461. - PubMed
-
- Foster SL, Hargreaves DC, Medzhitov R. Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature. 2007;447:972–978. - PubMed
-
- Garlanda C, Di Liberto D, Vecchi A, La Manna MP, Buracchi C, Caccamo N, Salerno A, Dieli F, Mantovani A. Damping excessive inflammation and tissue damage in Mycobacterium tuberculosis infection by Toll IL-1 receptor 8/single Ig IL-1-related receptor, a negative regulator of IL-1/TLR signaling. J Immunol. 2007;179:3119–3125. - PubMed
-
- Kawai T, Akira S. TLR signaling. Semin Immunol. 2007;19:24–32. - PubMed
-
- Lee MS, Kim YJ. Signaling pathways downstream of pattern-recognition receptors and their cross talk. Annu Rev Biochem. 2007;76:447–480. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
