

Pathophysiology of acute kidney injury
- PMID: 23798302
- PMCID: PMC3919808
- DOI: 10.1002/cphy.c110041
Pathophysiology of acute kidney injury
Abstract
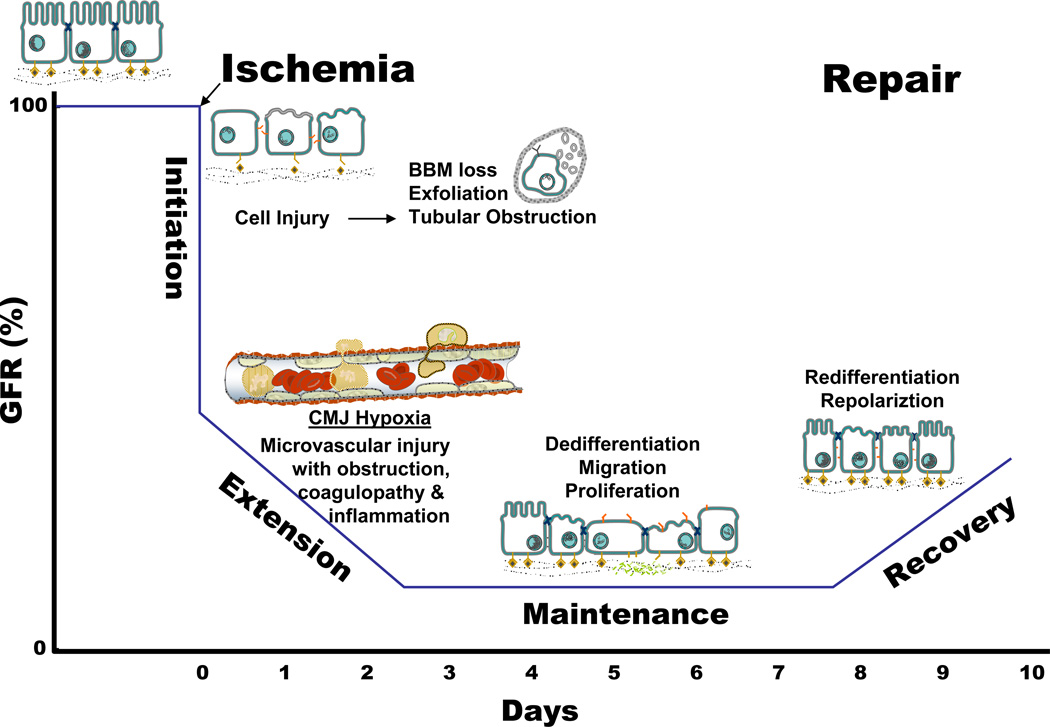
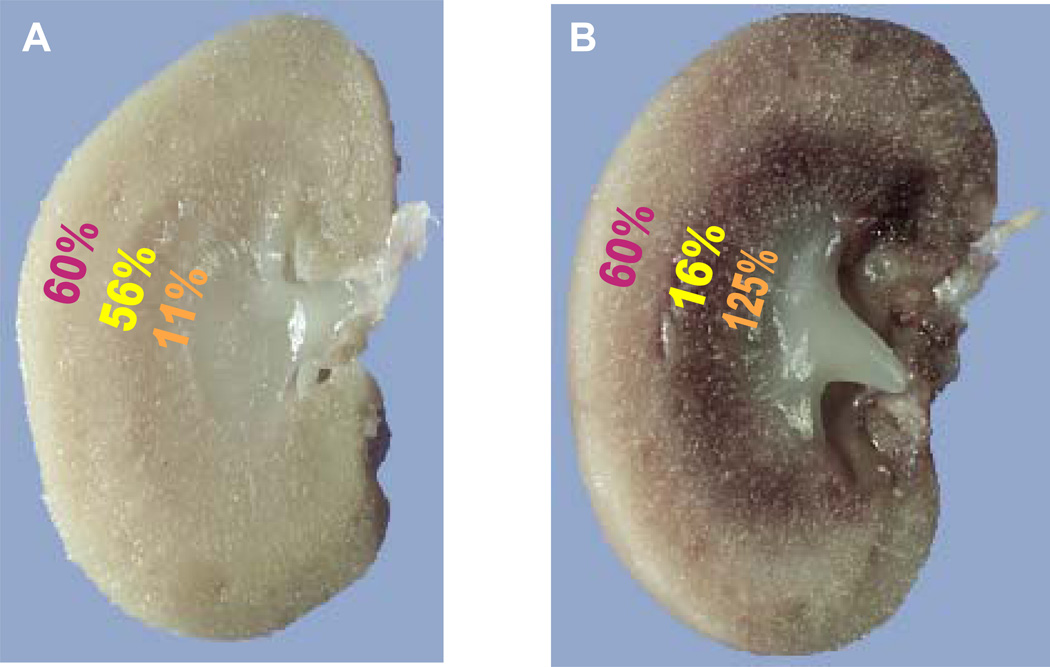
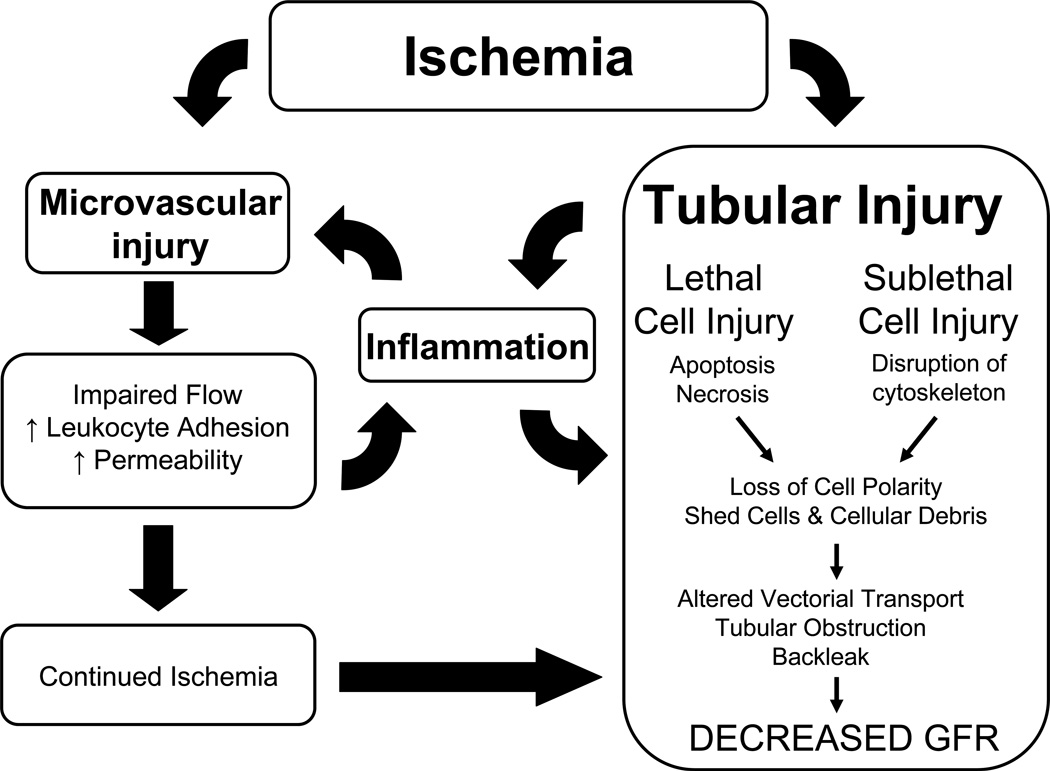
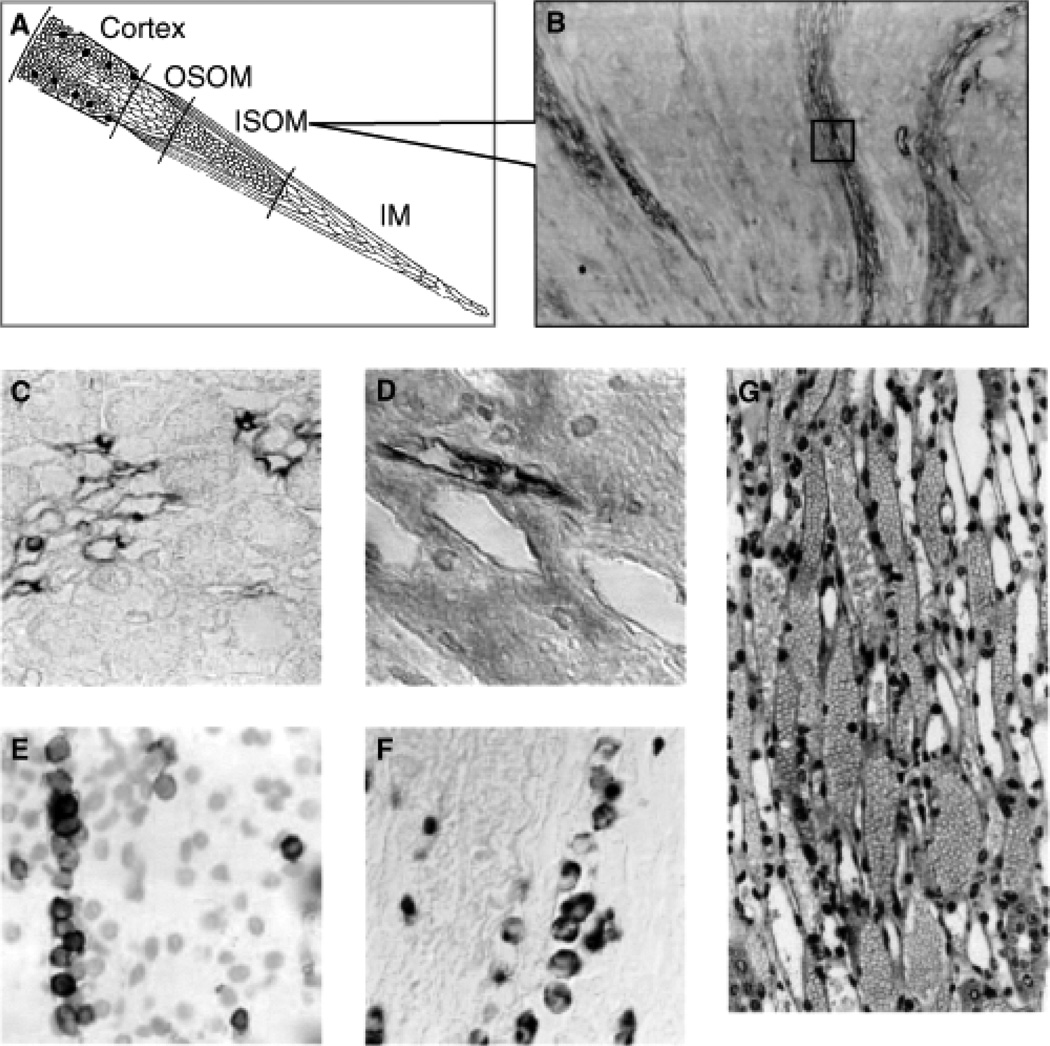
Acute kidney injury (AKI) is the leading cause of nephrology consultation and is associated with high mortality rates. The primary causes of AKI include ischemia, hypoxia, or nephrotoxicity. An underlying feature is a rapid decline in glomerular filtration rate (GFR) usually associated with decreases in renal blood flow. Inflammation represents an important additional component of AKI leading to the extension phase of injury, which may be associated with insensitivity to vasodilator therapy. It is suggested that targeting the extension phase represents an area potential of treatment with the greatest possible impact. The underlying basis of renal injury appears to be impaired energetics of the highly metabolically active nephron segments (i.e., proximal tubules and thick ascending limb) in the renal outer medulla, which can trigger conversion from transient hypoxia to intrinsic renal failure. Injury to kidney cells can be lethal or sublethal. Sublethal injury represents an important component in AKI, as it may profoundly influence GFR and renal blood flow. The nature of the recovery response is mediated by the degree to which sublethal cells can restore normal function and promote regeneration. The successful recovery from AKI depends on the degree to which these repair processes ensue and these may be compromised in elderly or chronic kidney disease (CKD) patients. Recent data suggest that AKI represents a potential link to CKD in surviving patients. Finally, earlier diagnosis of AKI represents an important area in treating patients with AKI that has spawned increased awareness of the potential that biomarkers of AKI may play in the future.
© 2012 American Physiological Society. Compr Physiol 2:1303-1353, 2012.
Figures



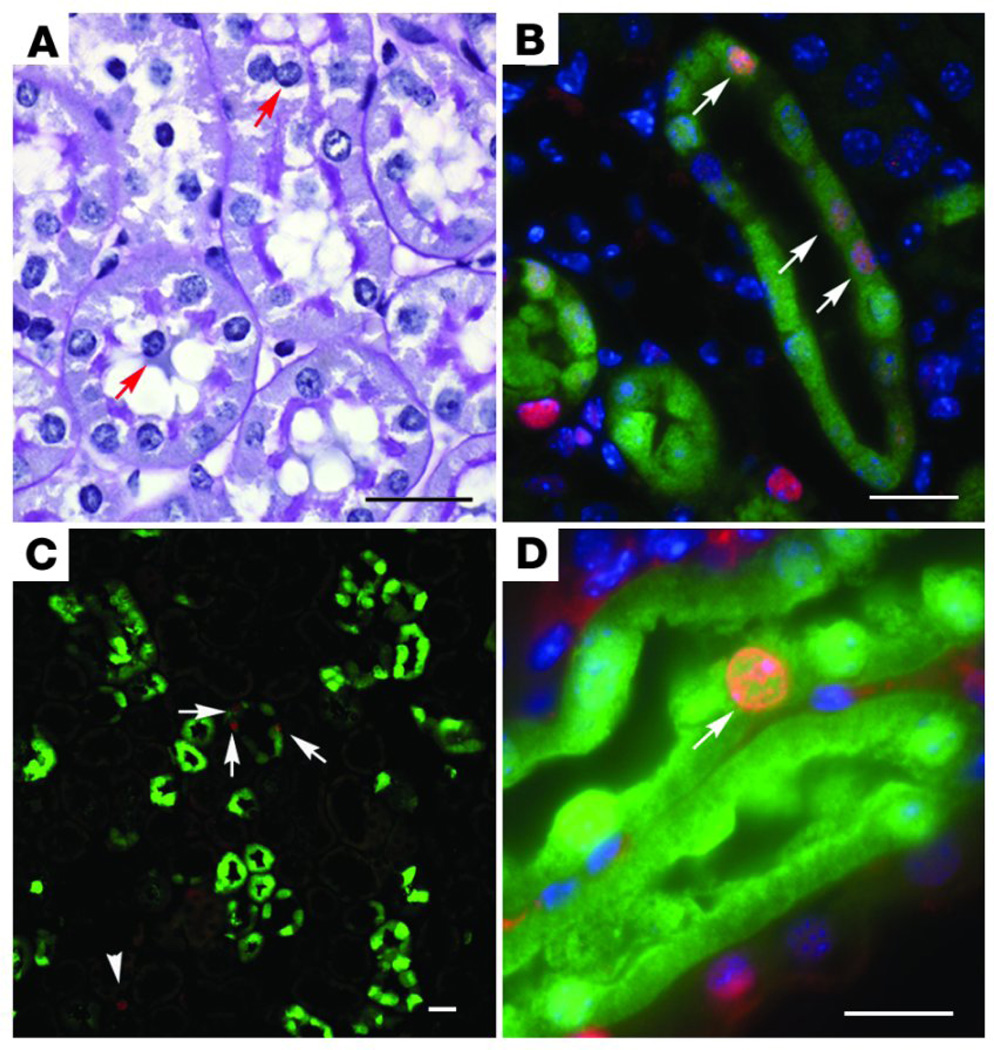
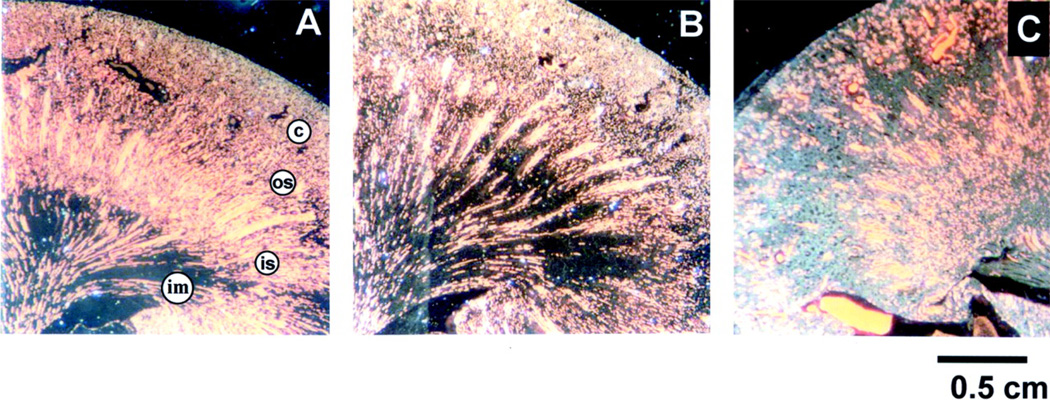
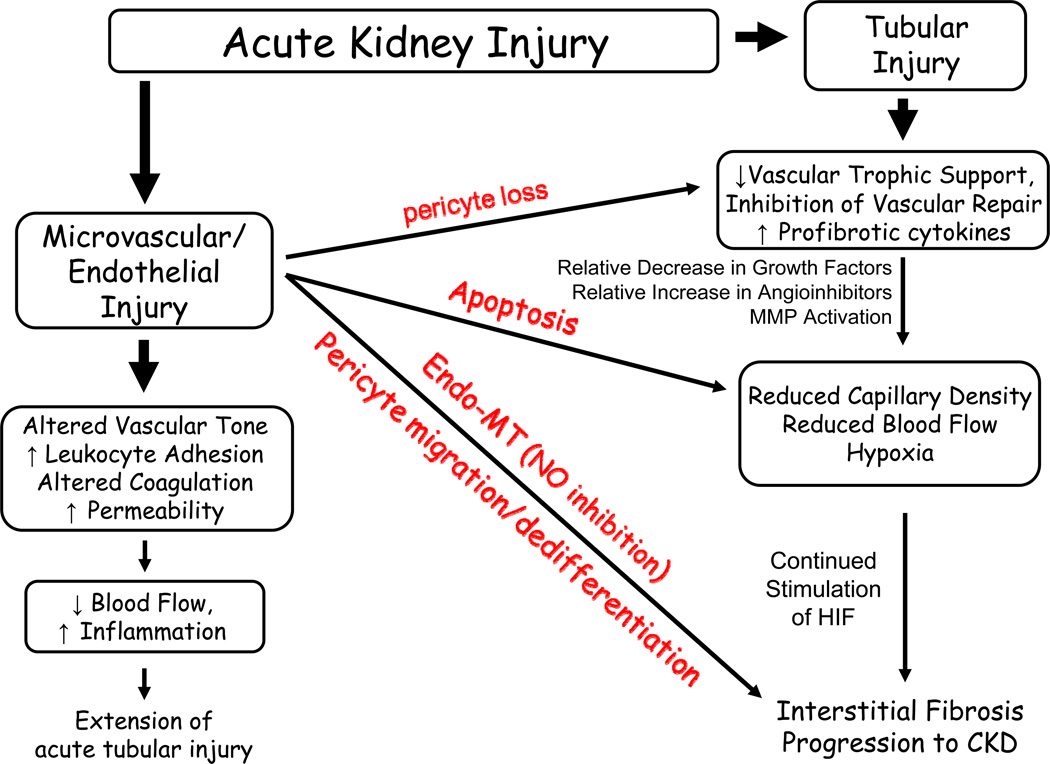
References
-
- Adabag AS, Ishani A, Koneswaran S, Johnson DJ, Kelly RF, Ward HB, McFalls EO, Bloomfield HE, Chandrashekhar Y. Utility of N-acetylcysteine to prevent acute kidney injury after cardiac surgery: A randomized controlled trial. American heart journal. 2008;155:1143–1149. - PubMed
-
- Agarwal A, Balla J, Alam J, Croatt AJ, Nath KA. Induction of heme oxygenase in toxic renal injury: A protective role in cisplatin nephrotoxicity in the rat. Kidney Int. 1995;48:1298–1307. - PubMed
-
- Aki Y, Nishiyama A, Miyatake A, Kimura S, Kohno M, Abe Y. Role of Adenosine A1 Receptor in Angiotensin II- and Norepinephrine-Induced Renal Vasoconstriction. Journal of Pharmacology and Experimental Therapeutics. 2002;303:117–123. - PubMed
-
- Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4:499–511. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
