

Region-based association analysis of human quantitative traits in related individuals
- PMID: 23799013
- PMCID: PMC3684601
- DOI: 10.1371/journal.pone.0065395
Region-based association analysis of human quantitative traits in related individuals
Abstract
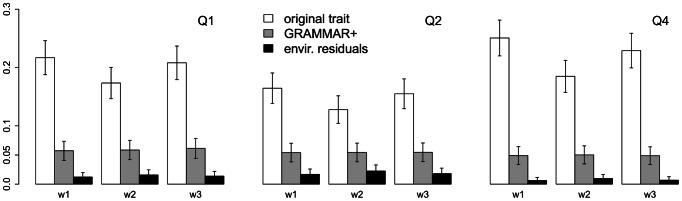
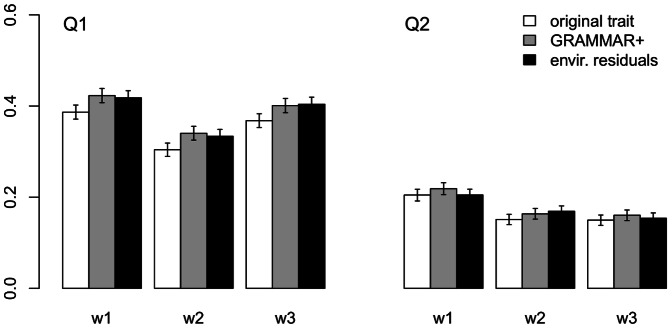
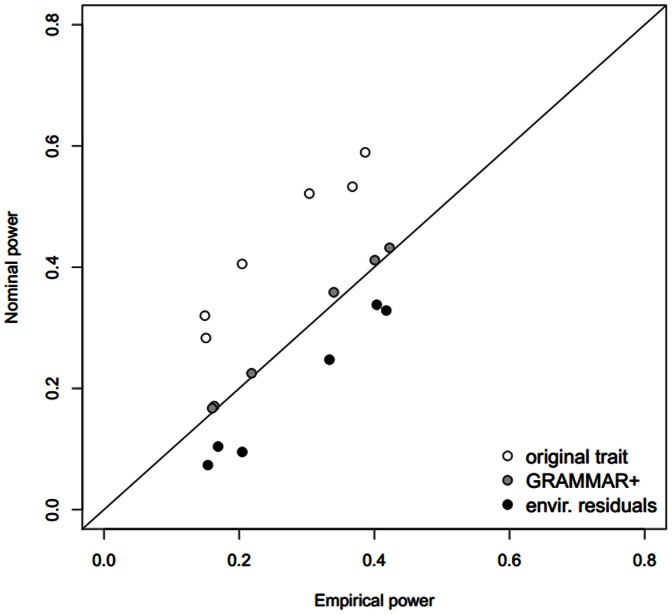
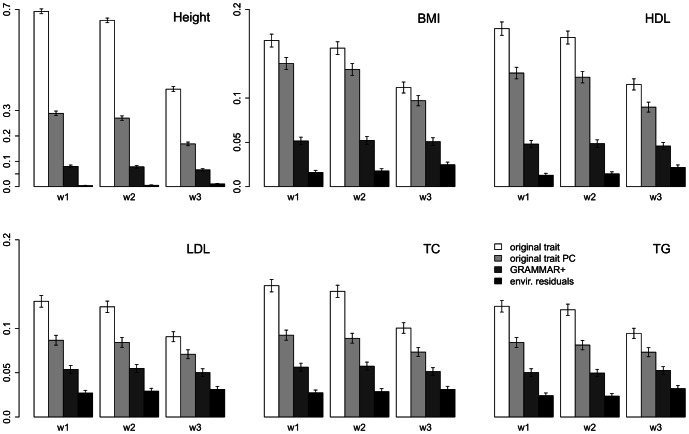
Regional-based association analysis instead of individual testing of each SNP was introduced in genome-wide association studies to increase the power of gene mapping, especially for rare genetic variants. For regional association tests, the kernel machine-based regression approach was recently proposed as a more powerful alternative to collapsing-based methods. However, the vast majority of existing algorithms and software for the kernel machine-based regression are applicable only to unrelated samples. In this paper, we present a new method for the kernel machine-based regression association analysis of quantitative traits in samples of related individuals. The method is based on the GRAMMAR+ transformation of phenotypes of related individuals, followed by use of existing kernel machine-based regression software for unrelated samples. We compared the performance of kernel-based association analysis on the material of the Genetic Analysis Workshop 17 family sample and real human data by using our transformation, the original untransformed trait, and environmental residuals. We demonstrated that only the GRAMMAR+ transformation produced type I errors close to the nominal value and that this method had the highest empirical power. The new method can be applied to analysis of related samples by using existing software for kernel-based association analysis developed for unrelated samples.
Conflict of interest statement
Figures
References
-
- Vineis P, Pearce N (2010) Missing heritability in genome-wide association study research. Nat Rev Genet 11: 589 doi: 10.1038/nrg2809-c2 - DOI - PubMed
-
- So HC, Gui AH, Cherny SS, Sham PC (2011) Evaluating the heritability explained by known susceptibility variants: a survey of ten complex diseases. Genet Epidemiol 35: 310–317. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
