

Mining breast cancer genes with a network based noise-tolerant approach
- PMID: 23799982
- PMCID: PMC3702465
- DOI: 10.1186/1752-0509-7-49
Mining breast cancer genes with a network based noise-tolerant approach
Abstract
Background: Mining novel breast cancer genes is an important task in breast cancer research. Many approaches prioritize candidate genes based on their similarity to known cancer genes, usually by integrating multiple data sources. However, different types of data often contain varying degrees of noise. For effective data integration, it's important to design methods that work robustly with respect to noise.
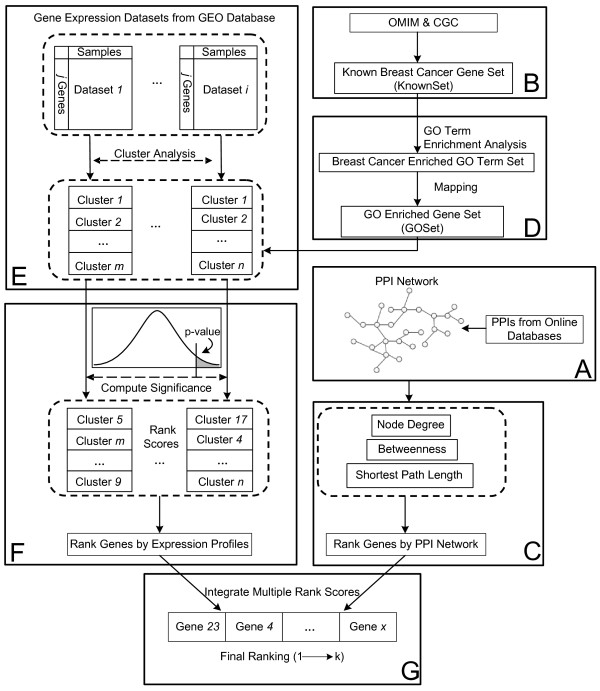
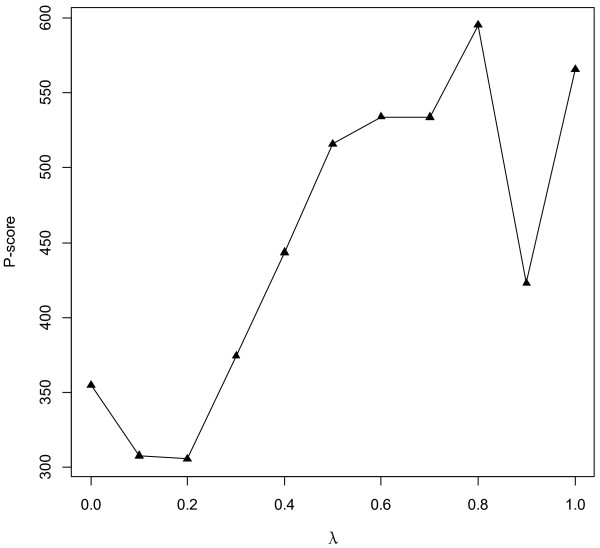
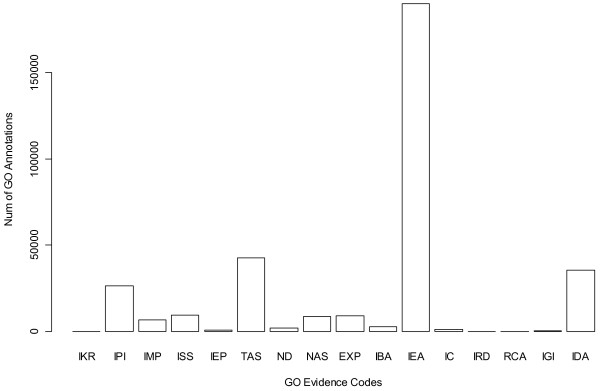
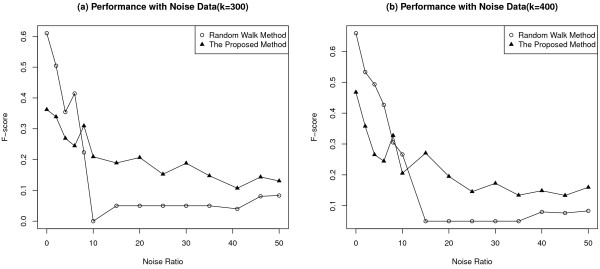
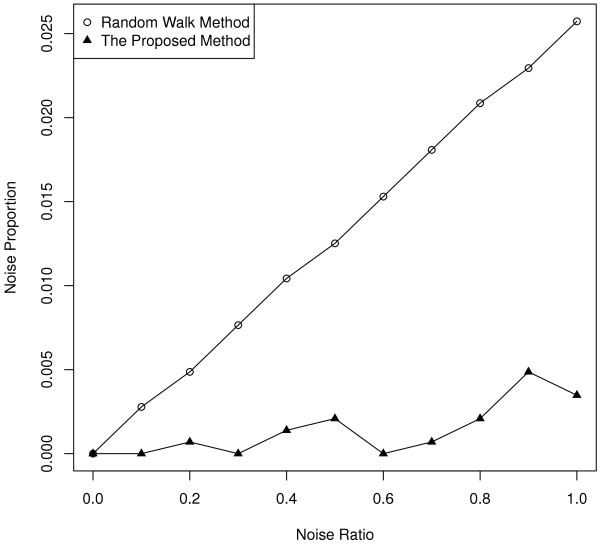
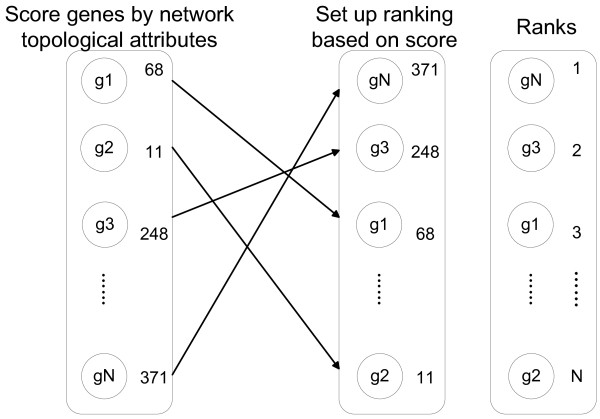
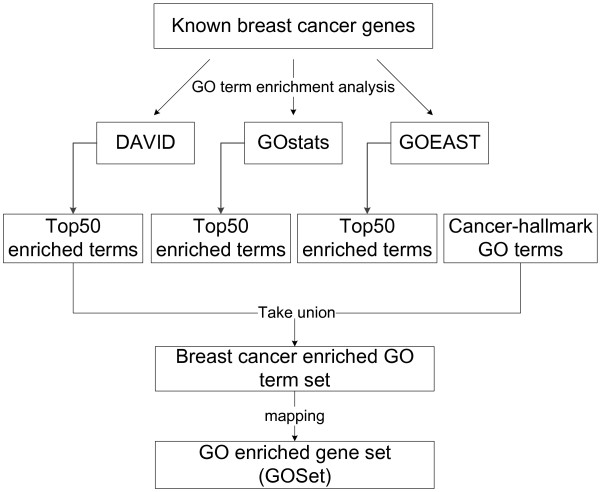
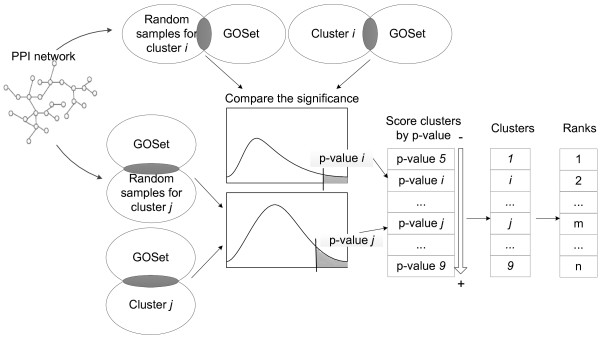
Results: Gene Ontology (GO) annotations were often utilized in cancer gene mining works. However, the vast majority of GO annotations were computationally derived, thus not completely accurate. A set of genes annotated with breast cancer enriched GO terms was adopted here as a set of source data with realistic noise. A novel noise tolerant approach was proposed to rank candidate breast cancer genes using noisy source data within the framework of a comprehensive human Protein-Protein Interaction (PPI) network. Performance of the proposed method was quantitatively evaluated by comparing it with the more established random walk approach. Results showed that the proposed method exhibited better performance in ranking known breast cancer genes and higher robustness against data noise than the random walk approach. When noise started to increase, the proposed method was able to maintained relatively stable performance, while the random walk approach showed drastic performance decline; when noise increased to a large extent, the proposed method was still able to achieve better performance than random walk did.
Conclusions: A novel noise tolerant method was proposed to mine breast cancer genes. Compared to the well established random walk approach, it showed better performance in correctly ranking cancer genes and worked robustly with respect to noise within source data. To the best of our knowledge, it's the first such effort to quantitatively analyze noise tolerance between different breast cancer gene mining methods. The sorted gene list can be valuable for breast cancer research. The proposed quantitative noise analysis method may also prove useful for other data integration efforts. It is hoped that the current work can lead to more discussions about influence of data noise on different computational methods for mining disease genes.
Figures
References
-
- Wu X, Li S. Cancer gene prediction using a network approach. Cancer Systems Biology. 2010. pp. 191–212.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
