

Prediction of gene-phenotype associations in humans, mice, and plants using phenologs
- PMID: 23800157
- PMCID: PMC3704650
- DOI: 10.1186/1471-2105-14-203
Prediction of gene-phenotype associations in humans, mice, and plants using phenologs
Abstract
Background: Phenotypes and diseases may be related to seemingly dissimilar phenotypes in other species by means of the orthology of underlying genes. Such "orthologous phenotypes," or "phenologs," are examples of deep homology, and may be used to predict additional candidate disease genes.
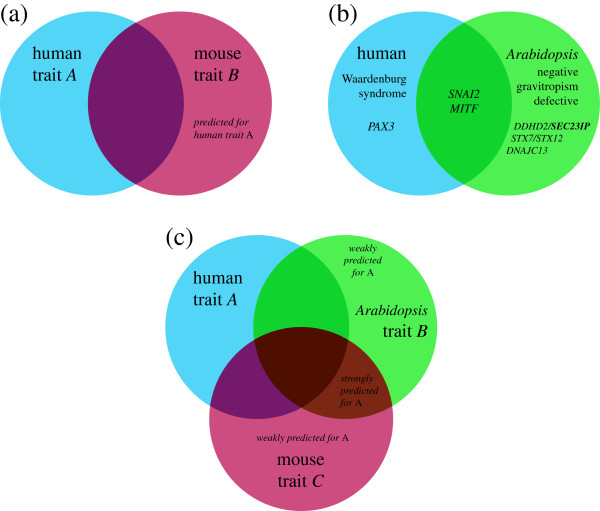
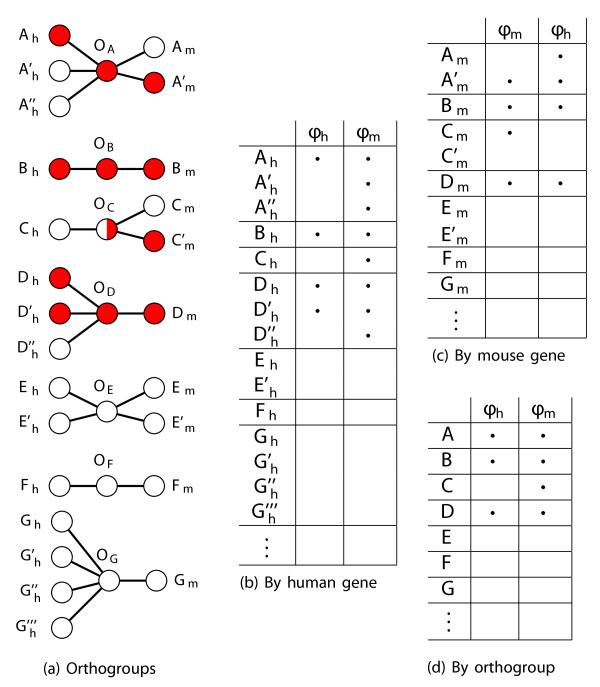
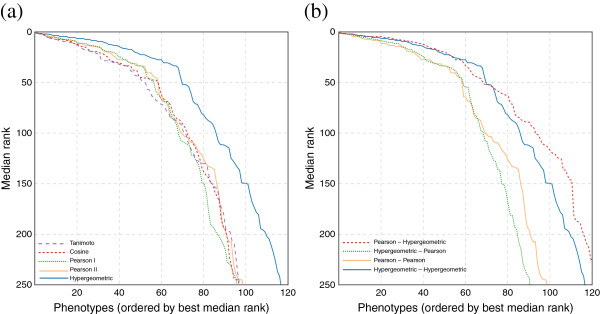
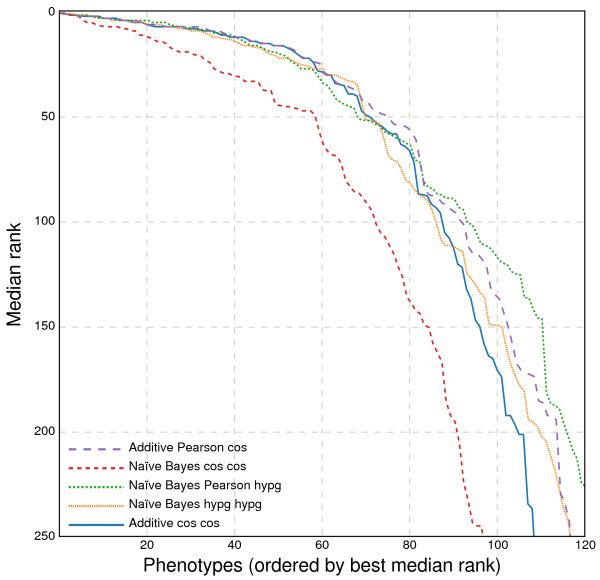
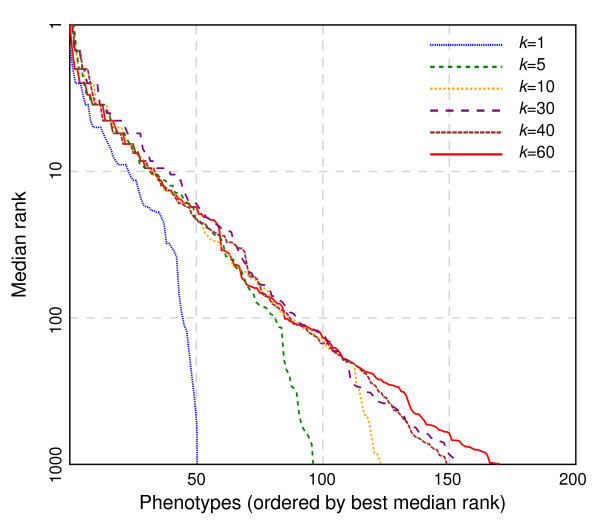
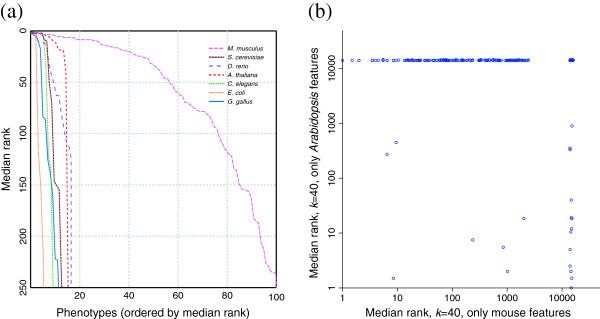
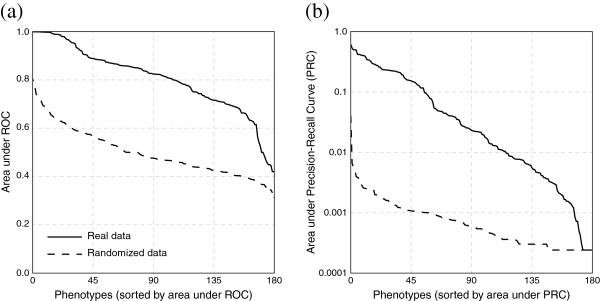
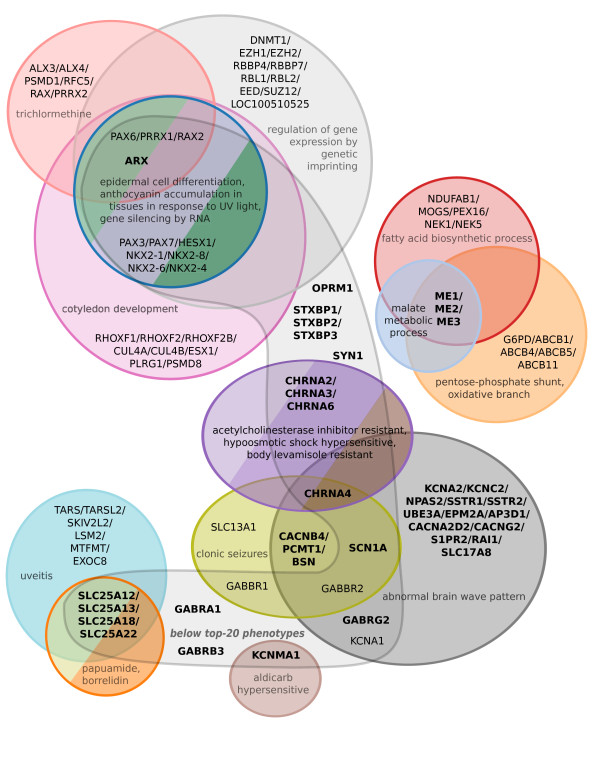
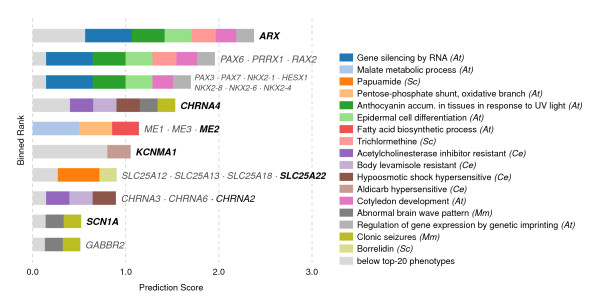
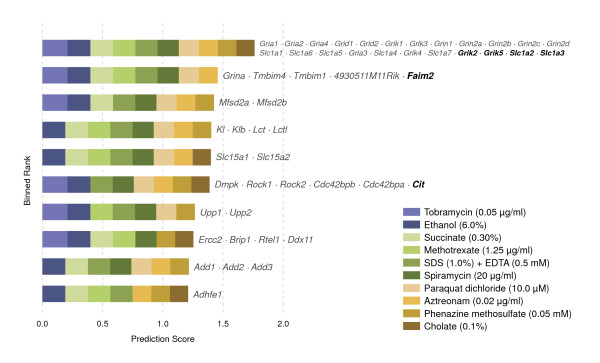
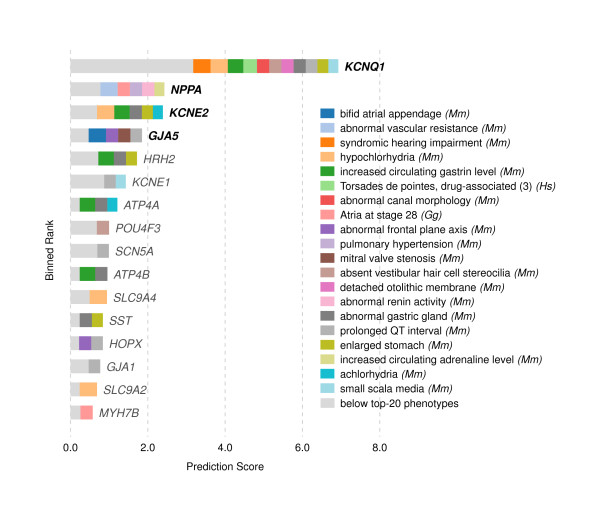
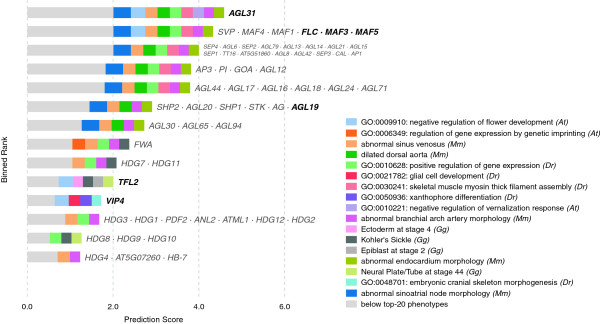
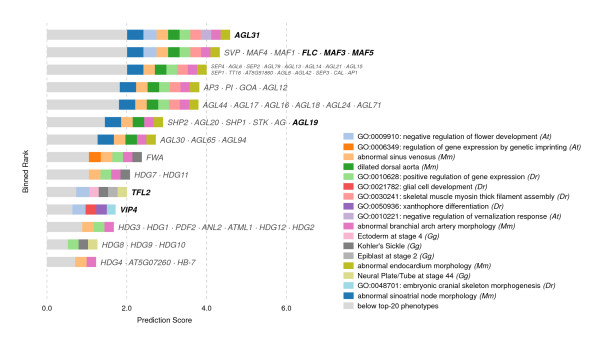
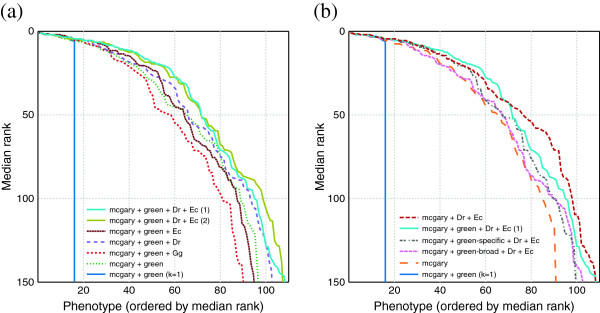
Results: In this work, we develop an unsupervised algorithm for ranking phenolog-based candidate disease genes through the integration of predictions from the k nearest neighbor phenologs, comparing classifiers and weighting functions by cross-validation. We also improve upon the original method by extending the theory to paralogous phenotypes. Our algorithm makes use of additional phenotype data--from chicken, zebrafish, and E. coli, as well as new datasets for C. elegans--establishing that several types of annotations may be treated as phenotypes. We demonstrate the use of our algorithm to predict novel candidate genes for human atrial fibrillation (such as HRH2, ATP4A, ATP4B, and HOPX) and epilepsy (e.g., PAX6 and NKX2-1). We suggest gene candidates for pharmacologically-induced seizures in mouse, solely based on orthologous phenotypes from E. coli. We also explore the prediction of plant gene-phenotype associations, as for the Arabidopsis response to vernalization phenotype.
Conclusions: We are able to rank gene predictions for a significant portion of the diseases in the Online Mendelian Inheritance in Man database. Additionally, our method suggests candidate genes for mammalian seizures based only on bacterial phenotypes and gene orthology. We demonstrate that phenotype information may come from diverse sources, including drug sensitivities, gene ontology biological processes, and in situ hybridization annotations. Finally, we offer testable candidates for a variety of human diseases, plant traits, and other classes of phenotypes across a wide array of species.
Figures
References
-
- Varma A, Palsson BO. Metabolic flux balancing: basic concepts, scientific and practical use. Nat Biotechnol. 1994;12:994–998. doi: 10.1038/nbt1094-994. - DOI
-
- Covert MW, Schilling CH, Palsson B. Regulation of gene expression in flux balance models of metabolism. J Theor Biol. 2001;213:73–88. doi: 10.1006/jtbi.2001.2405. [ http://www.ncbi.nlm.nih.gov/pubmed/11708855] - DOI - PubMed
-
- Covert M, Knight E, Reed J, Herrgard M, Palsson B. Integrating high-throughput and computational data elucidates bacterial networks. Nature. 2004;429(May):92–96. [ http://www.nature.com/nature/journal/v429/n6987/abs/nature02456.html] - PubMed
-
- Covert MW, Xiao N, Chen TJ, Karr JR. Integrating metabolic, transcriptional regulatory and signal transduction models in Escherichia coli. Bioinformatics (Oxford, England) 2008;24(18):2044–2050. doi: 10.1093/bioinformatics/btn352. [ http://www.ncbi.nlm.nih.gov/pubmed/18621757] - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
