

The million mutation project: a new approach to genetics in Caenorhabditis elegans
- PMID: 23800452
- PMCID: PMC3787271
- DOI: 10.1101/gr.157651.113
The million mutation project: a new approach to genetics in Caenorhabditis elegans
Abstract
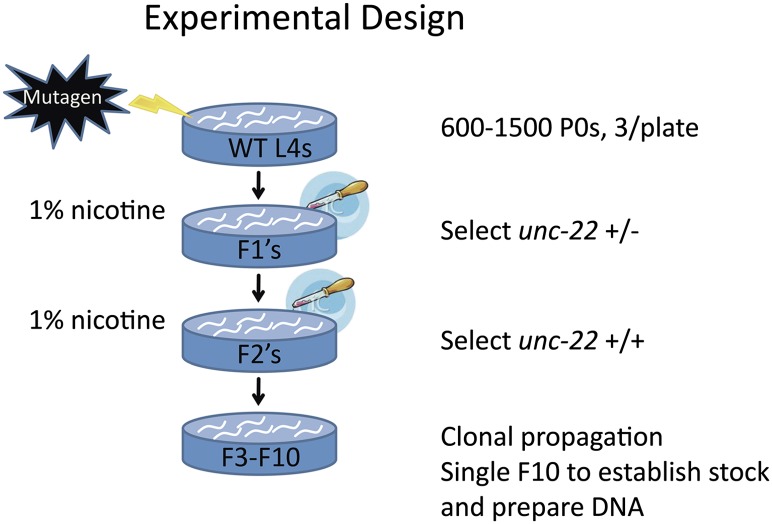
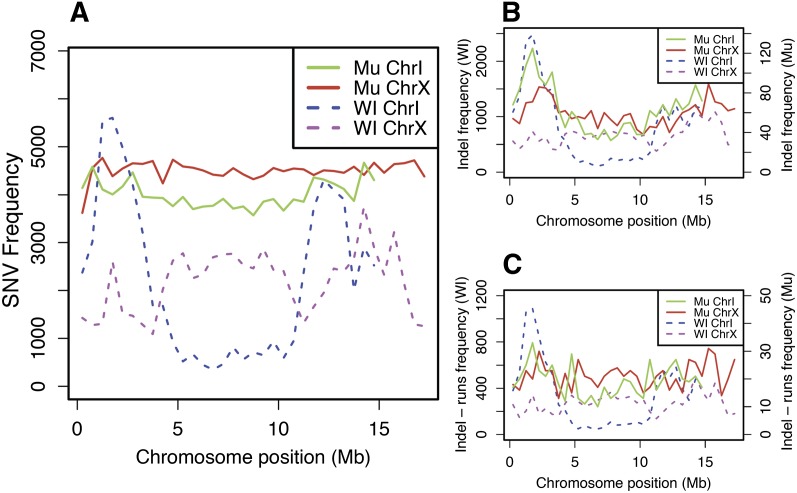
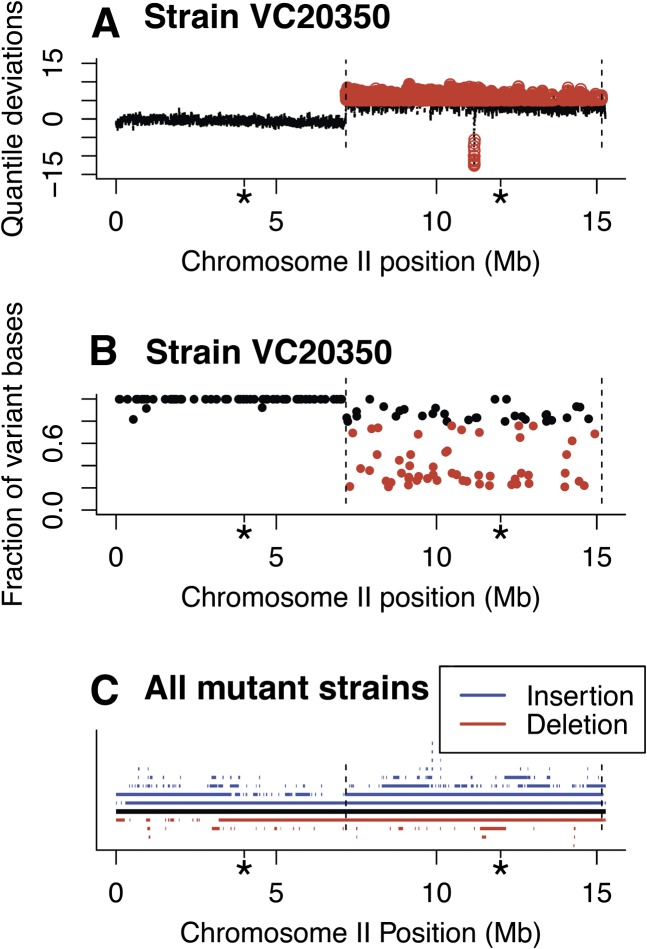
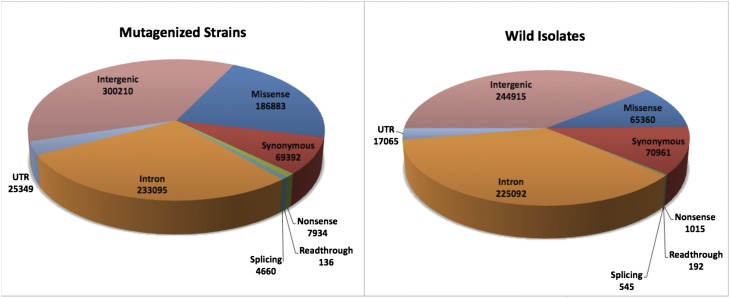
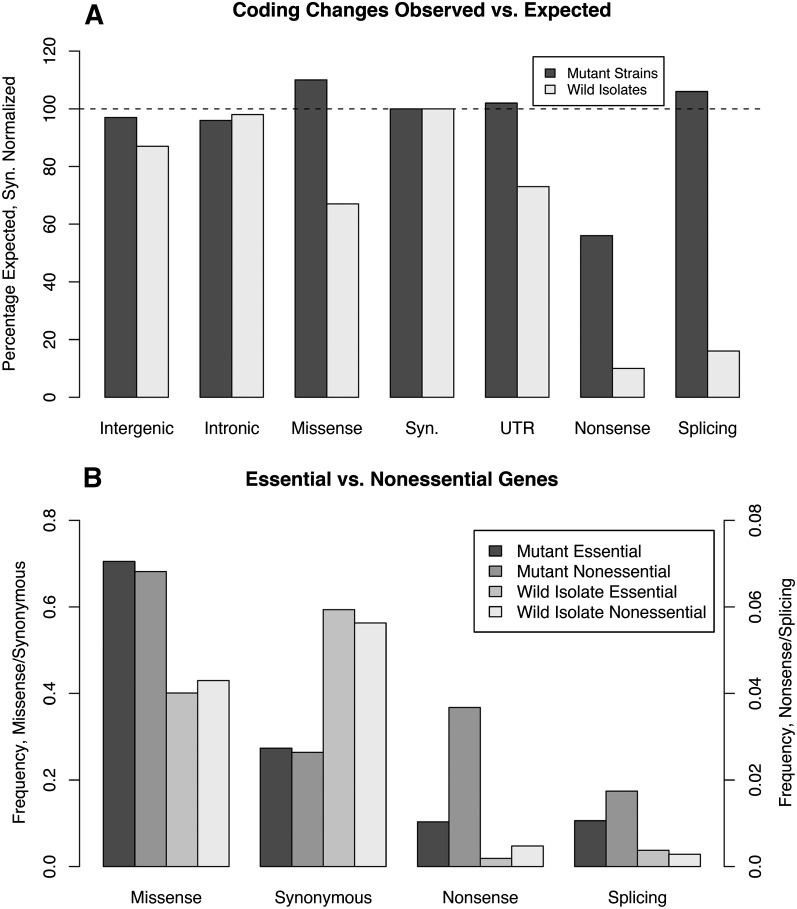
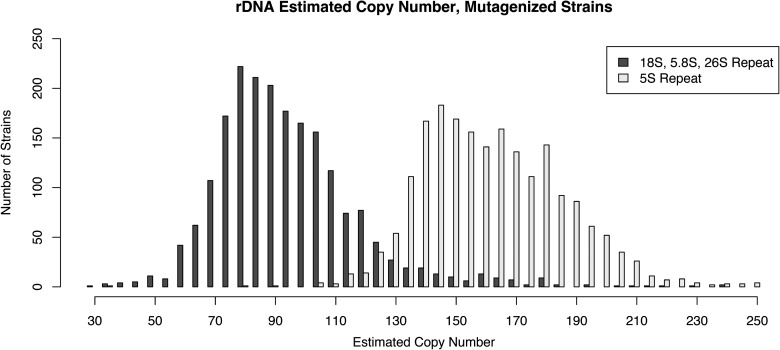
We have created a library of 2007 mutagenized Caenorhabditis elegans strains, each sequenced to a target depth of 15-fold coverage, to provide the research community with mutant alleles for each of the worm's more than 20,000 genes. The library contains over 800,000 unique single nucleotide variants (SNVs) with an average of eight nonsynonymous changes per gene and more than 16,000 insertion/deletion (indel) and copy number changes, providing an unprecedented genetic resource for this multicellular organism. To supplement this collection, we also sequenced 40 wild isolates, identifying more than 630,000 unique SNVs and 220,000 indels. Comparison of the two sets demonstrates that the mutant collection has a much richer array of both nonsense and missense mutations than the wild isolate set. We also find a wide range of rDNA and telomere repeat copy number in both sets. Scanning the mutant collection for molecular phenotypes reveals a nonsense suppressor as well as strains with higher levels of indels that harbor mutations in DNA repair genes and strains with abundant males associated with him mutations. All the strains are available through the Caenorhabditis Genetics Center and all the sequence changes have been deposited in WormBase and are available through an interactive website.
Figures
References
-
- Ahmed S, Hodgkin J 2000. MRT-2 checkpoint protein is required for germline immortality and telomere replication in C. elegans. Nature 403: 159–164 - PubMed
-
- Bailey JA, Gu Z, Clark RA, Reinert K, Samonte RV, Schwartz S, Adams MD, Myers EW, Li PW, Eichler EE 2002. Recent segmental duplications in the human genome. Science 297: 1003–1007 - PubMed
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources