

Sulindac selectively inhibits colon tumor cell growth by activating the cGMP/PKG pathway to suppress Wnt/β-catenin signaling
- PMID: 23804703
- PMCID: PMC3800150
- DOI: 10.1158/1535-7163.MCT-13-0048
Sulindac selectively inhibits colon tumor cell growth by activating the cGMP/PKG pathway to suppress Wnt/β-catenin signaling
Abstract
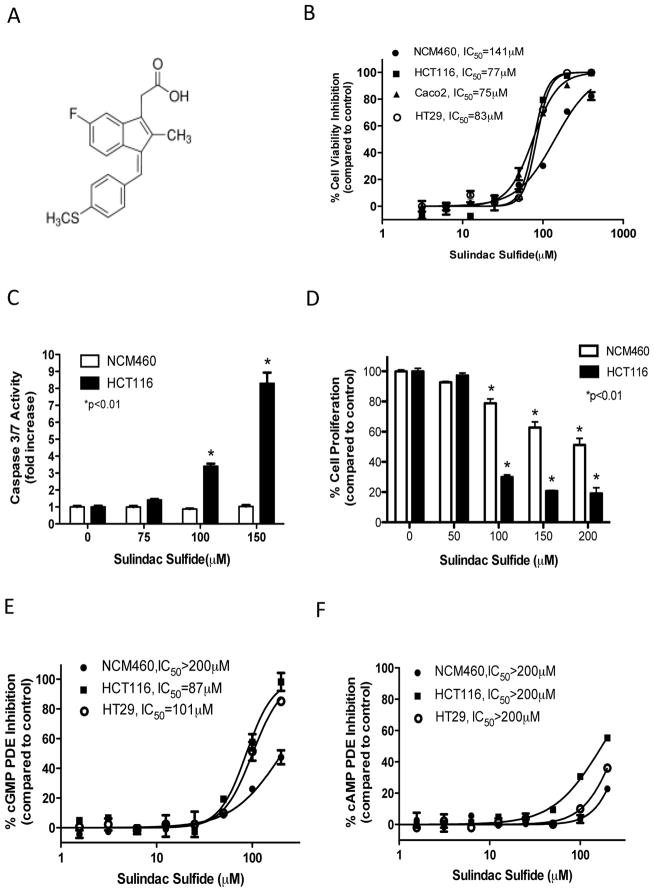
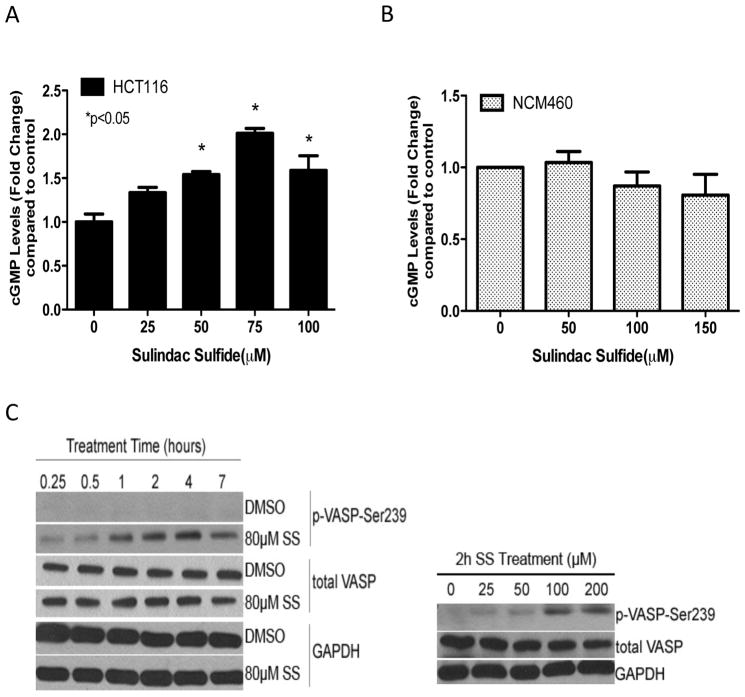
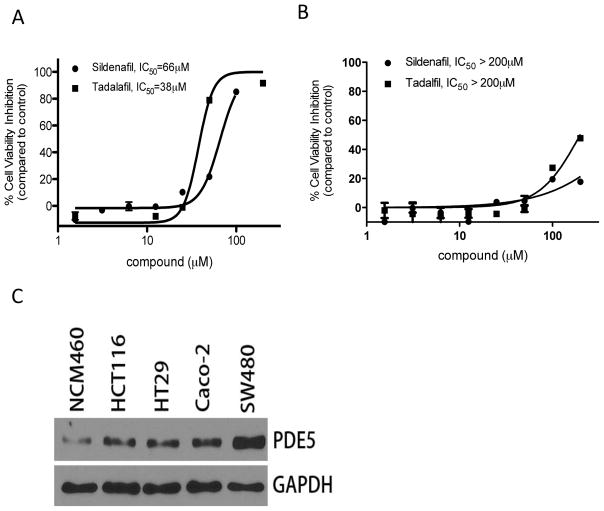
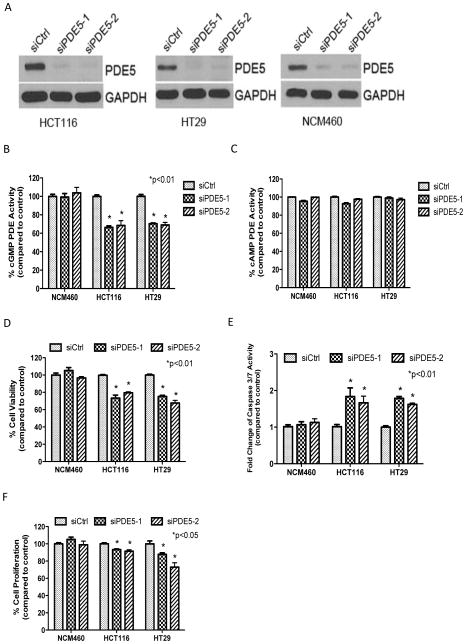
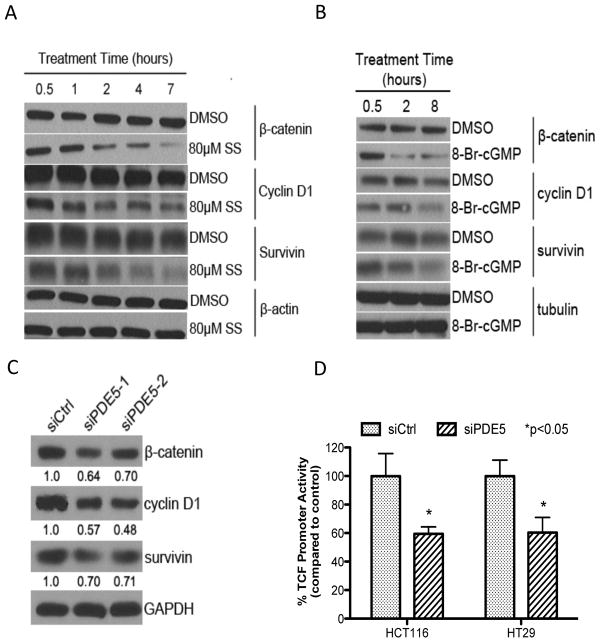
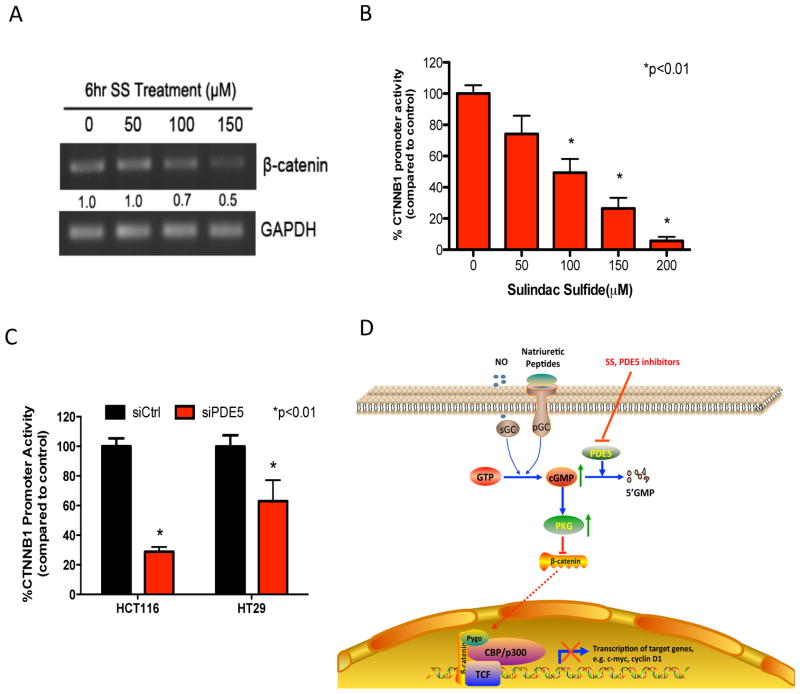
Nonsteroidal anti-inflammatory drugs (NSAID) display promising antineoplastic activity for colorectal and other cancers, but toxicity from COX inhibition limits their long-term use for chemoprevention. Previous studies have concluded that the basis for their tumor cell growth inhibitory activity does not require COX inhibition, although the underlying mechanism is poorly understood. Here, we report that the NSAID sulindac sulfide inhibits cyclic guanosine 3',5'-monophosphate phosphodiesterase (cGMP PDE) activity to increase intracellular cGMP levels and activate cGMP-dependent protein kinase (PKG) at concentrations that inhibit proliferation and induce apoptosis of colon tumor cells. Sulindac sulfide did not activate the cGMP/PKG pathway, nor affect proliferation or apoptosis in normal colonocytes. Knockdown of the cGMP-specific PDE5 isozyme by siRNA and PDE5-specific inhibitors tadalafil and sildenafil also selectively inhibited the growth of colon tumor cells that expressed high levels of PDE5 compared with colonocytes. The mechanism by which sulindac sulfide and the cGMP/PKG pathway inhibits colon tumor cell growth involves the transcriptional suppression of β-catenin to inhibit Wnt/β-catenin T-cell factor transcriptional activity, leading to downregulation of cyclin D1 and survivin. These observations suggest that safer and more efficacious sulindac derivatives can be developed for colorectal cancer chemoprevention by targeting PDE5 and possibly other cGMP-degrading isozymes.
Conflict of interest statement
Figures
References
-
- Jemal A, Murray T, Ward E, Samuels A, Tiwari RC, Ghafoor A, et al. Cancer statistics, 2005. CA Cancer J Clin. 2005;55:10–30. - PubMed
-
- Wingo PA, Ries LA, Parker SL, Heath CW., Jr Long-term cancer patient survival in the United States. Cancer Epidemiol Biomarkers Prev. 1998;7:271–82. - PubMed
-
- Greenlee RT, Hill-Harmon MB, Murray T, Thun M. Cancer statistics, 2001. CA Cancer J Clin. 2001;51:15–36. - PubMed
-
- Chan TA. Nonsteroidal anti-inflammatory drugs, apoptosis, and colon-cancer chemoprevention. Lancet Oncol. 2002;3:166–74. - PubMed
-
- Thun MJ, Henley SJ, Patrono C. Nonsteroidal anti-inflammatory drugs as anticancer agents: mechanistic, pharmacologic, and clinical issues. J Natl Cancer Inst. 2002;94:252–66. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
