

Ribosome profiling provides evidence that large noncoding RNAs do not encode proteins
- PMID: 23810193
- PMCID: PMC3756563
- DOI: 10.1016/j.cell.2013.06.009
Ribosome profiling provides evidence that large noncoding RNAs do not encode proteins
Abstract
Large noncoding RNAs are emerging as an important component in cellular regulation. Considerable evidence indicates that these transcripts act directly as functional RNAs rather than through an encoded protein product. However, a recent study of ribosome occupancy reported that many large intergenic ncRNAs (lincRNAs) are bound by ribosomes, raising the possibility that they are translated into proteins. Here, we show that classical noncoding RNAs and 5' UTRs show the same ribosome occupancy as lincRNAs, demonstrating that ribosome occupancy alone is not sufficient to classify transcripts as coding or noncoding. Instead, we define a metric based on the known property of translation whereby translating ribosomes are released upon encountering a bona fide stop codon. We show that this metric accurately discriminates between protein-coding transcripts and all classes of known noncoding transcripts, including lincRNAs. Taken together, these results argue that the large majority of lincRNAs do not function through encoded proteins.
Copyright © 2013 Elsevier Inc. All rights reserved.
Figures
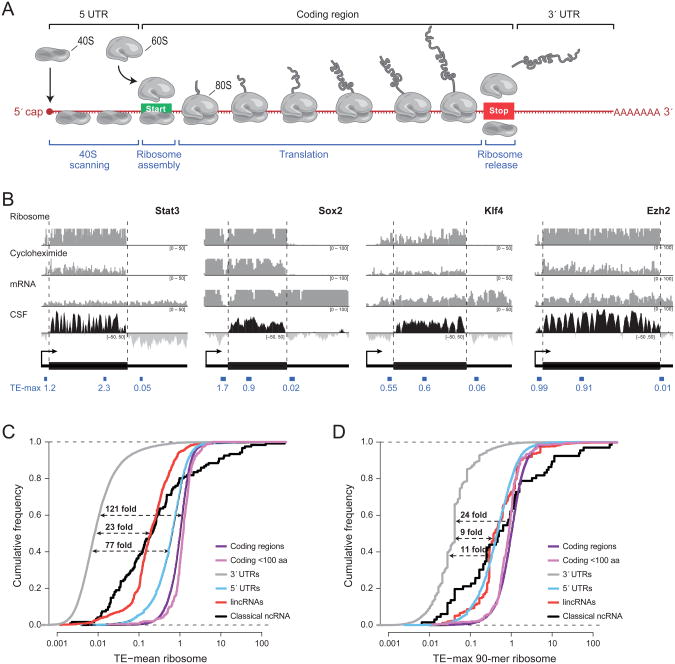
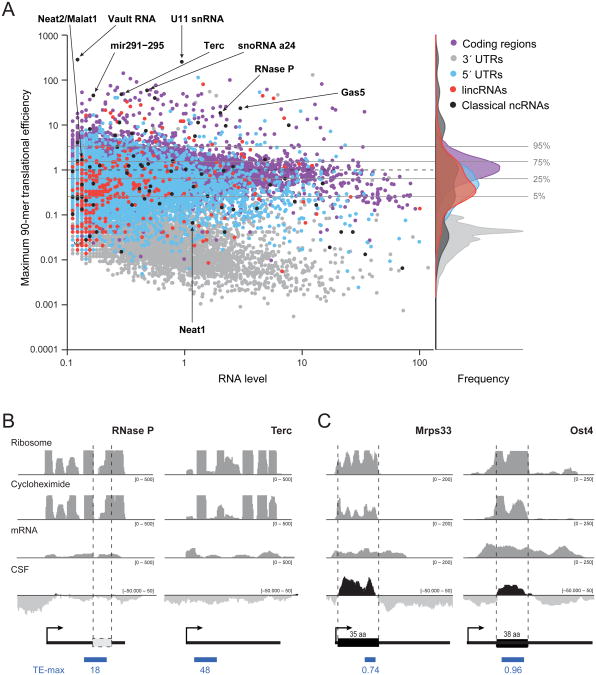
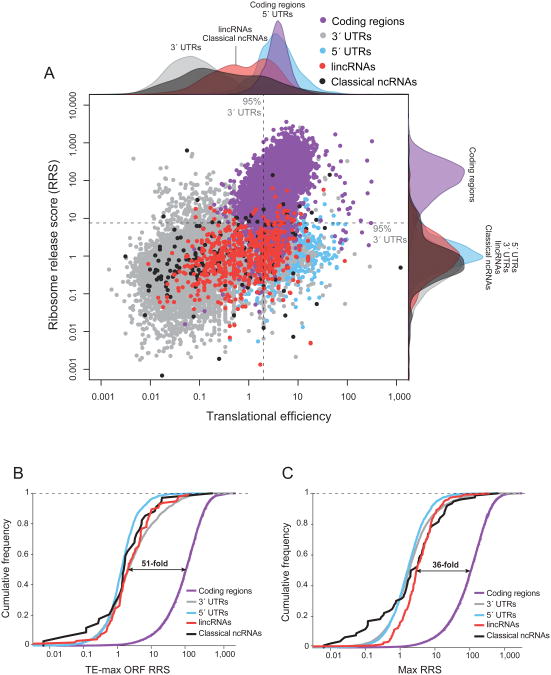
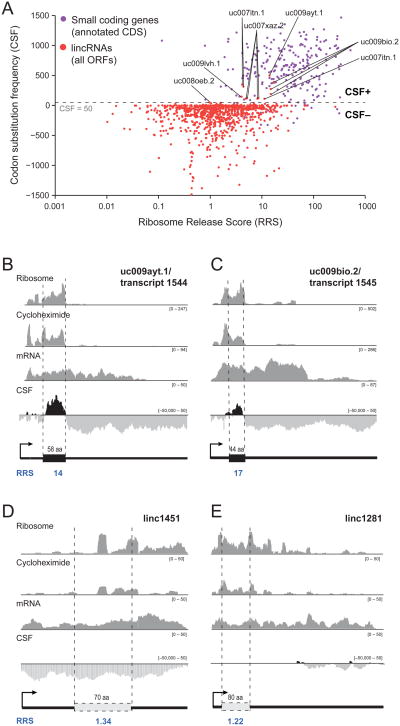
Comment in
-
Non-coding RNA: Ribosomes, but no translation, for lincRNAs.Nat Rev Genet. 2013 Aug;14(8):520. doi: 10.1038/nrg3534. Epub 2013 Jul 9. Nat Rev Genet. 2013. PMID: 23835437 No abstract available.
References
-
- Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. - PubMed
-
- Carninci P, Kasukawa T, Katayama S, Gough J, Frith MC, Maeda N, Oyama R, Ravasi T, Lenhard B, Wells C, et al. The transcriptional landscape of the mammalian genome. Science. 2005;309:1559–1563. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
