

CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation
- PMID: 23812099
- PMCID: PMC3720827
- DOI: 10.1038/ni.2639
CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation
Abstract
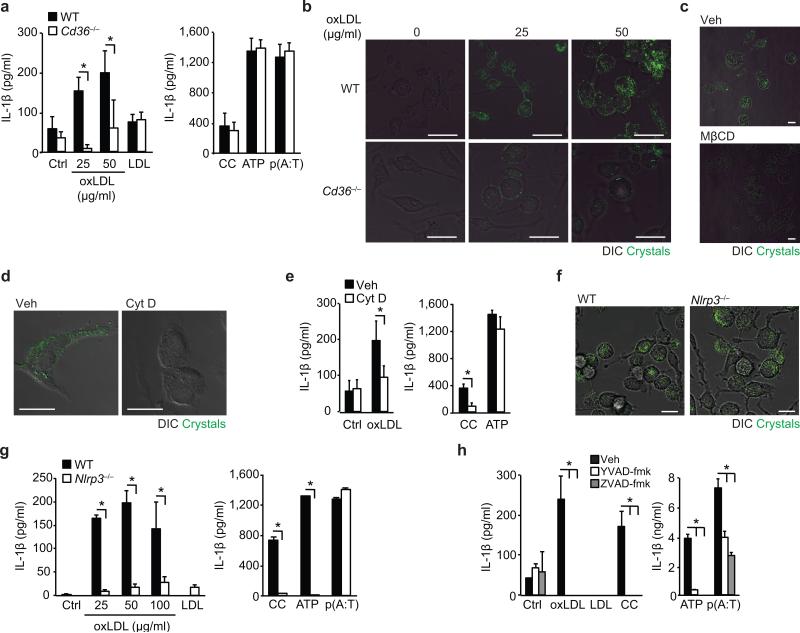
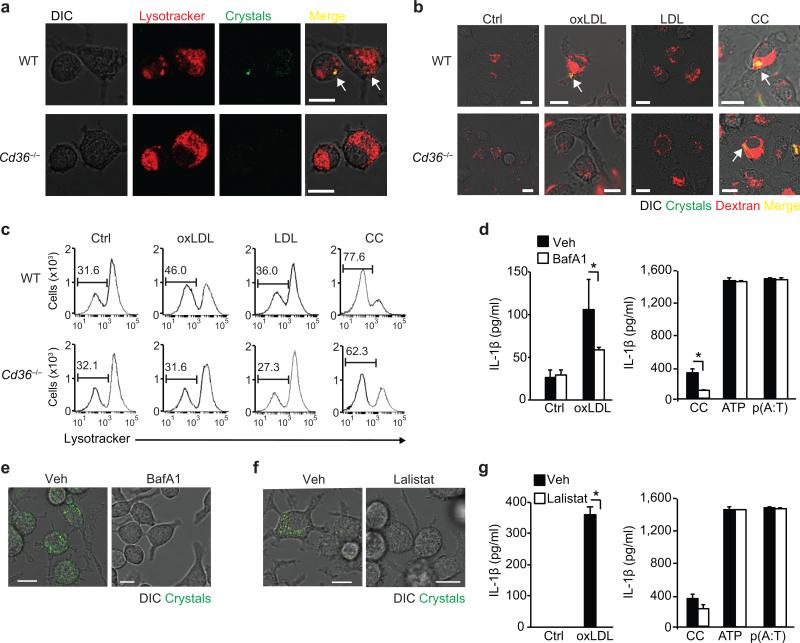
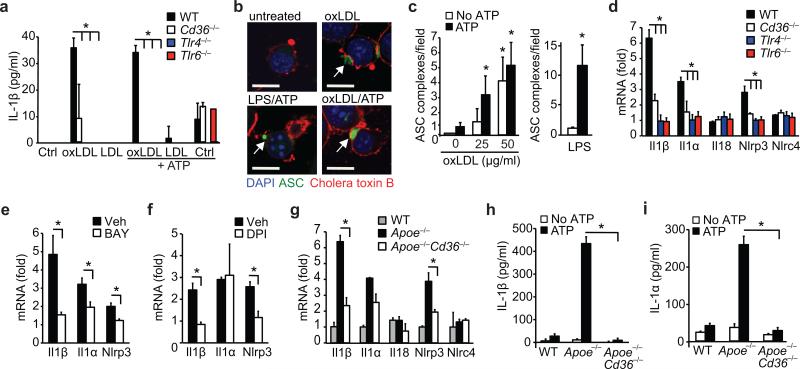
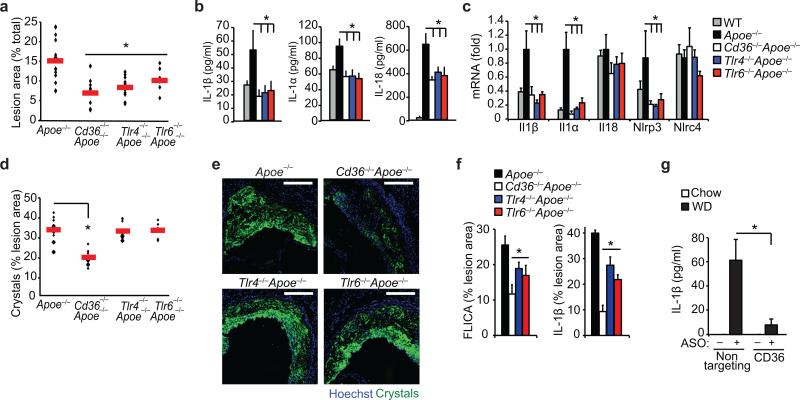
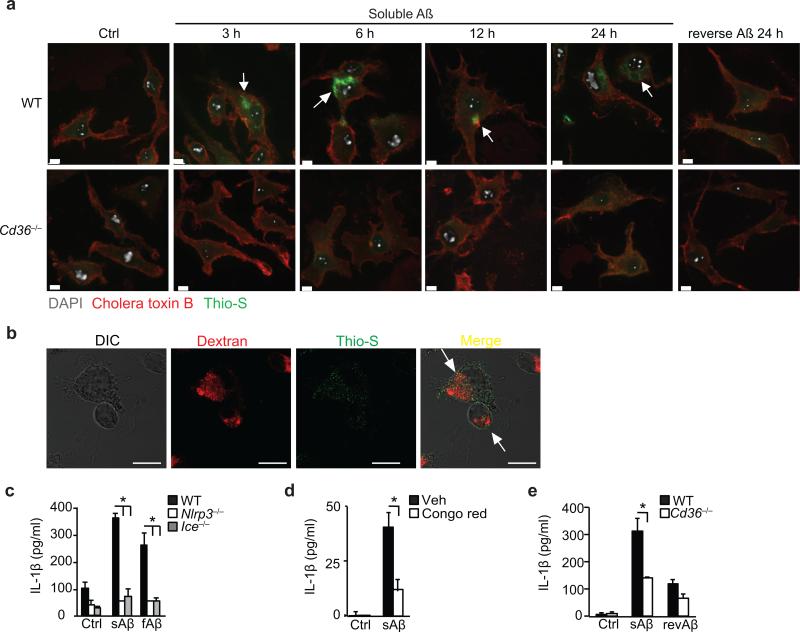
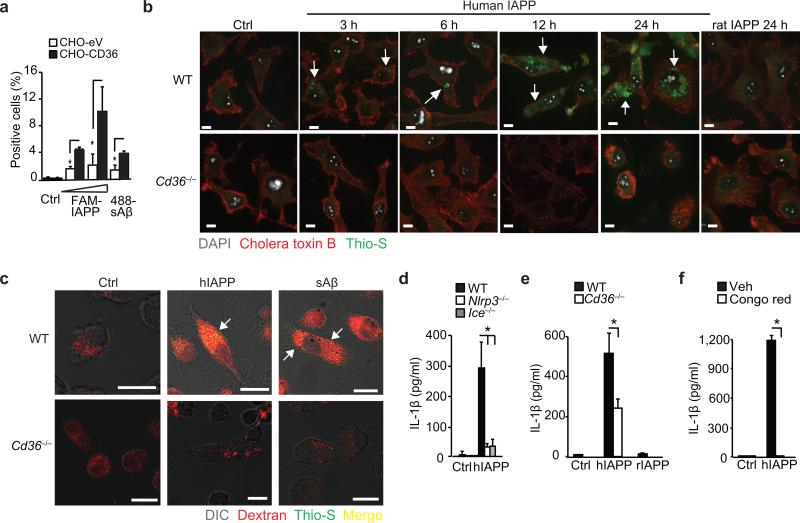
Particulate ligands, including cholesterol crystals and amyloid fibrils, induce production of interleukin 1β (IL-1β) dependent on the cytoplasmic sensor NLRP3 in atherosclerosis, Alzheimer's disease and diabetes. Soluble endogenous ligands, including oxidized low-density lipoprotein (LDL), amyloid-β and amylin peptides, accumulate in such diseases. Here we identify an endocytic pathway mediated by the pattern-recognition receptor CD36 that coordinated the intracellular conversion of those soluble ligands into crystals or fibrils, which resulted in lysosomal disruption and activation of the NLRP3 inflammasome. Consequently, macrophages that lacked CD36 failed to elicit IL-1β production in response to those ligands, and targeting CD36 in atherosclerotic mice resulted in lower serum concentrations of IL-1β and accumulation of cholesterol crystals in plaques. Collectively, our findings highlight the importance of CD36 in the accrual and nucleation of NLRP3 ligands from within the macrophage and position CD36 as a central regulator of inflammasome activation in sterile inflammation.
Figures
Comment in
-
NLRP3 inflammasome activation: CD36 serves double duty.Nat Immunol. 2013 Aug;14(8):772-4. doi: 10.1038/ni.2668. Nat Immunol. 2013. PMID: 23867926 Free PMC article.
-
CD36: linking lipids to the NLRP3 inflammasome, atherogenesis and atherothrombosis.Cell Mol Immunol. 2014 Jan;11(1):8-10. doi: 10.1038/cmi.2013.48. Epub 2013 Oct 7. Cell Mol Immunol. 2014. PMID: 24097033 Free PMC article. No abstract available.
References
-
- Abela GS. Cholesterol crystals piercing the arterial plaque and intima trigger local and systemic inflammation. J. Clin. Lipidol. 2010;4:156–164. - PubMed
-
- Westermark GT, Westermark P, Berne C, Korsgren O. Nordic Network for Clinical Islet T. Widespread amyloid deposition in transplanted human pancreatic islets. N. Engl. J. Med. 2008;359:977–979. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- P30 DK043351/DK/NIDDK NIH HHS/United States
- R01 HL117334/HL/NHLBI NIH HHS/United States
- R01 AG032349/AG/NIA NIH HHS/United States
- 1R01HL112661-01/HL/NHLBI NIH HHS/United States
- U24 AI082660/AI/NIAID NIH HHS/United States
- R01 HL093262/HL/NHLBI NIH HHS/United States
- R01HL117334/HL/NHLBI NIH HHS/United States
- R01 AI083713/AI/NIAID NIH HHS/United States
- AI083713/AI/NIAID NIH HHS/United States
- R01AG032349/AG/NIA NIH HHS/United States
- 5R01HL093262-02/HL/NHLBI NIH HHS/United States
- R01 HL112661/HL/NHLBI NIH HHS/United States
- R01 AI079198/AI/NIAID NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
