

Strategies for optimizing the response of cancer and normal tissues to radiation
- PMID: 23812271
- PMCID: PMC3906736
- DOI: 10.1038/nrd4003
Strategies for optimizing the response of cancer and normal tissues to radiation
Abstract
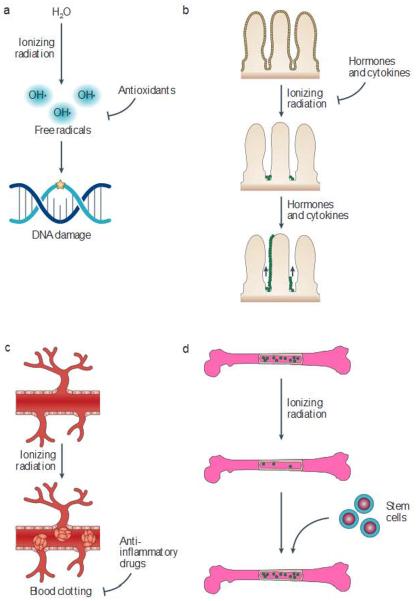
Approximately 50% of all patients with cancer receive radiation therapy at some point during the course of their treatment, and the majority of these patients are treated with curative intent. Despite recent advances in the planning of radiation treatment and the delivery of image-guided radiation therapy, acute toxicity and potential long-term side effects often limit the ability to deliver a sufficient dose of radiation to control tumours locally. In the past two decades, a better understanding of the hallmarks of cancer and the discovery of specific signalling pathways by which cells respond to radiation have provided new opportunities to design molecularly targeted therapies to increase the therapeutic window of radiation therapy. Here, we review efforts to develop approaches that could improve outcomes with radiation therapy by increasing the probability of tumour cure or by decreasing normal tissue toxicity.
Figures
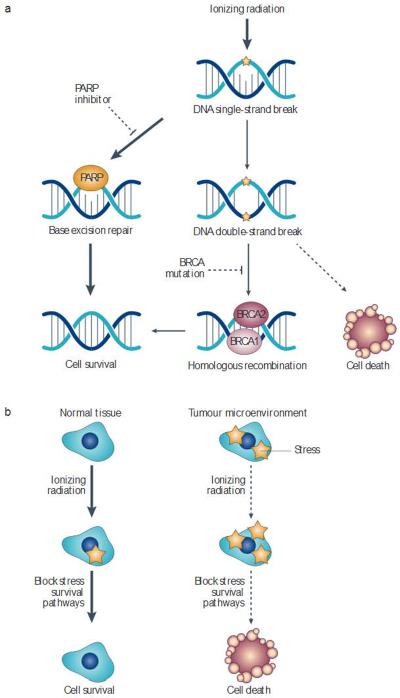
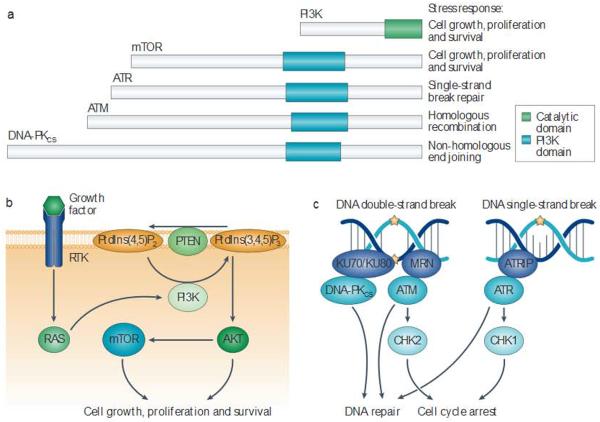
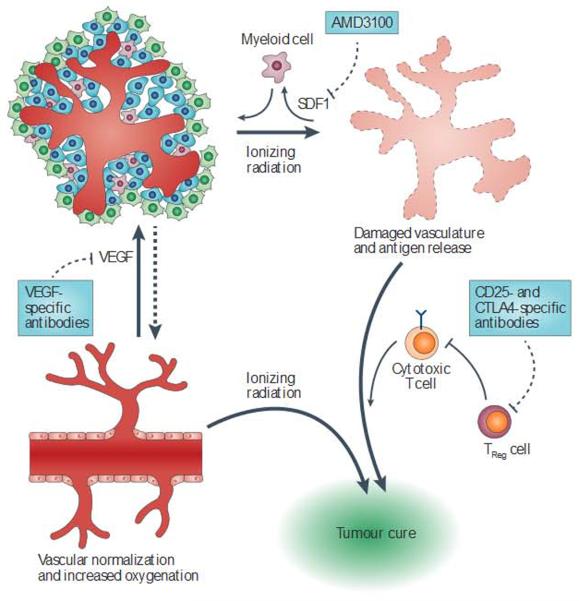
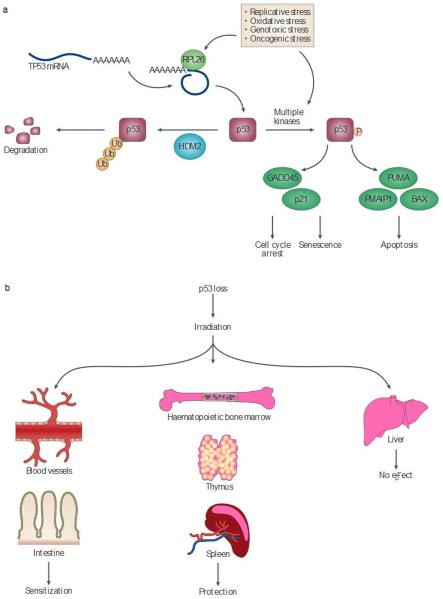
References
-
- Intensity Modulated Radiation Therapy Collaborative Working Group Intensity-modulated radiotherapy: current status and issues of interest. Int. J. Radiat. Oncol. Biol. Phys. 2001;51:880–914. - PubMed
-
- Lo SS, et al. Stereotactic body radiation therapy: a novel treatment modality. Nature Rev. Clin. Oncol. 2010;7:44–54. - PubMed
-
This manuscript reviews prospective clinical trials-and current clinical use of stereotactic body radiation therapy.
-
- Timmerman R, et al. Excessive toxicity when treating central tumors in a phase II study of stereotactic body radiation therapy for medically inoperable early-stage lung cancer. J. Clin. Oncol. 2006;24:4835–4839. - PubMed
-
- Forquer JA, et al. Brachial plexopathy from stereotactic body radiotherapy in early-stage NSCLC: dose-limiting toxicity In apical tumor sites. Radiother. Oncol. 2009;93:408–413. - PubMed
-
- Sahgal A, Larson DA, Chang EL. Stereotactic body radiosurgery for spinal metastases: a critical review. Int. J. Radiat Oncol. Biol. Phys. 2008;71:652–665. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
