

FGFR1 mutations cause Hartsfield syndrome, the unique association of holoprosencephaly and ectrodactyly
- PMID: 23812909
- PMCID: PMC3756455
- DOI: 10.1136/jmedgenet-2013-101603
FGFR1 mutations cause Hartsfield syndrome, the unique association of holoprosencephaly and ectrodactyly
Abstract
Background: Harstfield syndrome is the rare and unique association of holoprosencephaly (HPE) and ectrodactyly, with or without cleft lip and palate, and variable additional features. All the reported cases occurred sporadically. Although several causal genes of HPE and ectrodactyly have been identified, the genetic cause of Hartsfield syndrome remains unknown. We hypothesised that a single key developmental gene may underlie the co-occurrence of HPE and ectrodactyly.
Methods: We used whole exome sequencing in four isolated cases including one case-parents trio, and direct Sanger sequencing of three additional cases, to investigate the causative variants in Hartsfield syndrome.
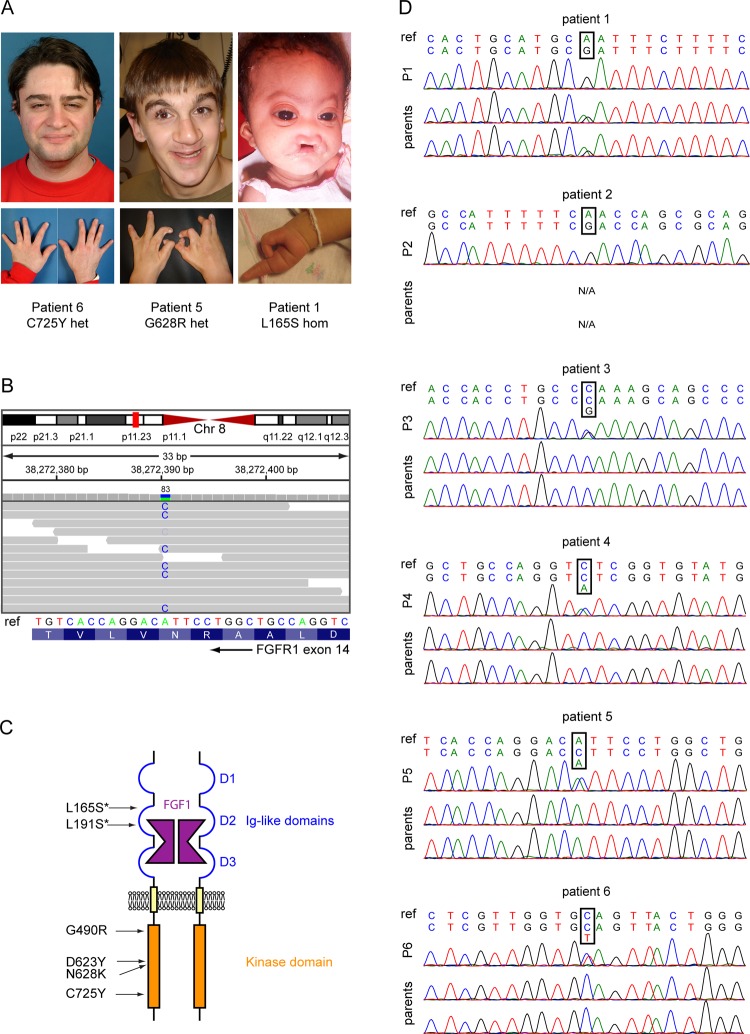
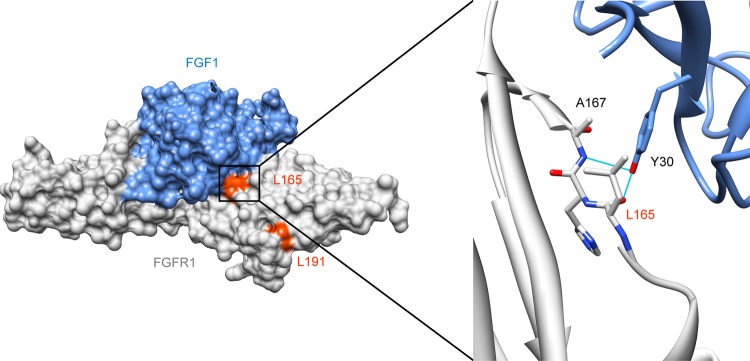
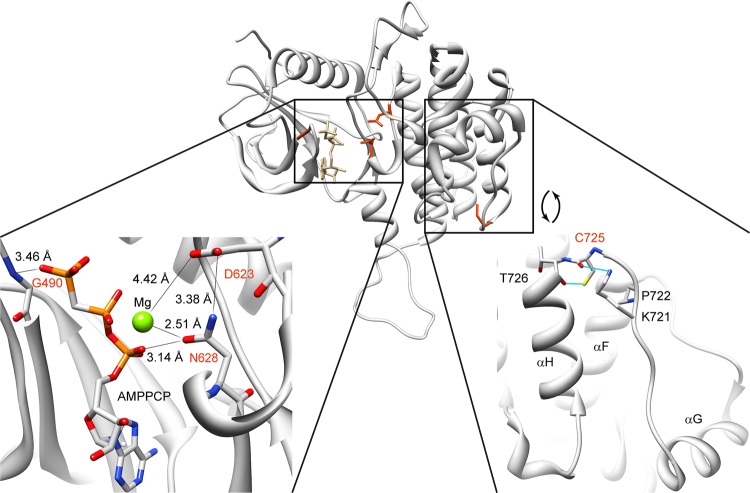
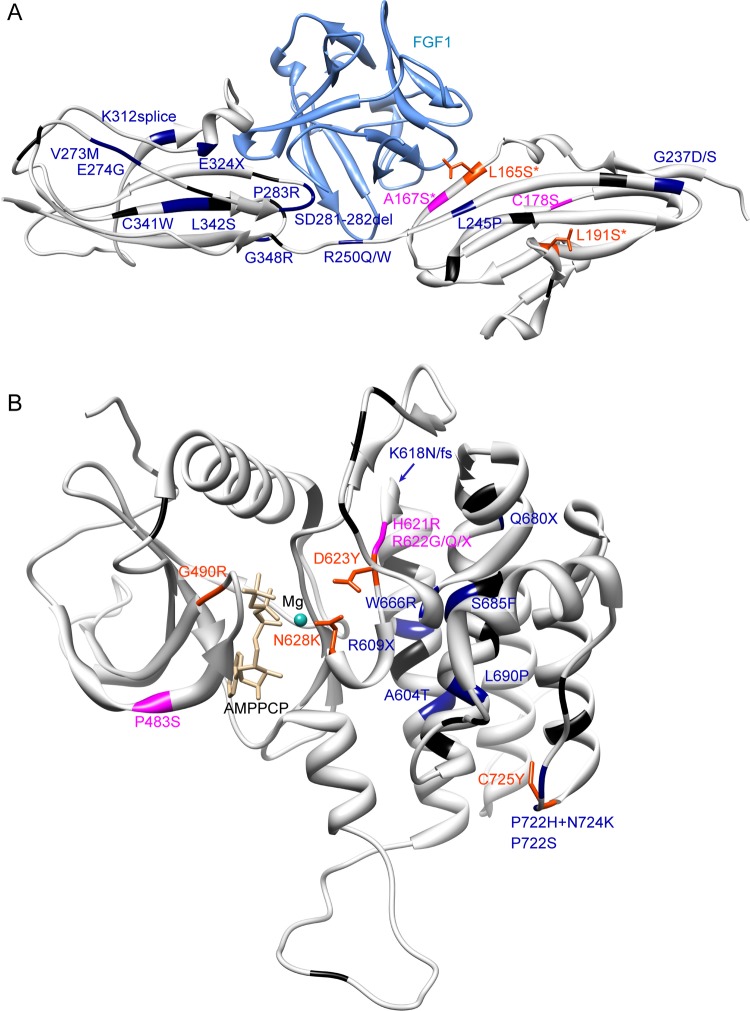
Results: We identified a novel FGFR1 mutation in six out of seven patients. Affected residues are highly conserved and are located in the extracellular binding domain of the receptor (two homozygous mutations) or the intracellular tyrosine kinase domain (four heterozygous de novo variants). Strikingly, among the six novel mutations, three are located in close proximity to the ATP's phosphates or the coordinating magnesium, with one position required for kinase activity, and three are adjacent to known mutations involved in Kallmann syndrome plus other developmental anomalies.
Conclusions: Dominant or recessive FGFR1 mutations are responsible for Hartsfield syndrome, consistent with the known roles of FGFR1 in vertebrate ontogeny and conditional Fgfr1-deficient mice. Our study shows that, in humans, lack of accurate FGFR1 activation can disrupt both brain and hand/foot midline development, and that FGFR1 loss-of-function mutations are responsible for a wider spectrum of clinical anomalies than previously thought, ranging in severity from seemingly isolated hypogonadotropic hypogonadism, through Kallmann syndrome with or without additional features, to Hartsfield syndrome at its most severe end.
Keywords: Clinical genetics; Developmental; Genetics.
Figures
References
-
- Cohen MM., Jr Holoprosencephaly: clinical, anatomic, and molecular dimensions. Birth Defects Res A Clin Mol Teratol 2006;76:658–73 - PubMed
-
- Duijf PH, van Bokhoven H, Brunner HG. Pathogenesis of split-hand/split-foot malformation. Hum Mol Genet 2003;12(Spec No 1):R51–60 - PubMed
-
- de Mollerat XJ, Gurrieri F, Morgan CT, Sangiorgi E, Everman DB, Gaspari P, Amiel J, Bamshad MJ, Lyle R, Blouin JL, Allanson JE, Le Marec B, Wilson M, Braverman NE, Radhakrishna U, Delozier-Blanchet C, Abbott A, Elghouzzi V, Antonarakis S, Stevenson RE, Munnich A, Neri G, Schwartz CE. A genomic rearrangement resulting in a tandem duplication is associated with split hand-split foot malformation 3 (SHFM3) at 10q24. Hum Mol Genet 2003;12:1959–71 - PubMed
-
- Hartsfield J, Bixler D, DeMeyer W. Hypertelorism associated with holoprosencephaly and ectrodactyly. J Clin Dysmorphol 1984:27–31
-
- Vilain C, Mortier G, Van Vliet G, Dubourg C, Heinrichs C, de Silva D, Verloes A, Baumann C. Hartsfield holoprosencephaly-ectrodactyly syndrome in five male patients: further delineation and review. Am J Med Genet A 2009;149A:1476–81 - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous