

Short read alignment with populations of genomes
- PMID: 23813006
- PMCID: PMC3694645
- DOI: 10.1093/bioinformatics/btt215
Short read alignment with populations of genomes
Abstract
Summary: The increasing availability of high-throughput sequencing technologies has led to thousands of human genomes having been sequenced in the past years. Efforts such as the 1000 Genomes Project further add to the availability of human genome variation data. However, to date, there is no method that can map reads of a newly sequenced human genome to a large collection of genomes. Instead, methods rely on aligning reads to a single reference genome. This leads to inherent biases and lower accuracy. To tackle this problem, a new alignment tool BWBBLE is introduced in this article. We (i) introduce a new compressed representation of a collection of genomes, which explicitly tackles the genomic variation observed at every position, and (ii) design a new alignment algorithm based on the Burrows-Wheeler transform that maps short reads from a newly sequenced genome to an arbitrary collection of two or more (up to millions of) genomes with high accuracy and no inherent bias to one specific genome.
Availability: http://viq854.github.com/bwbble.
Figures
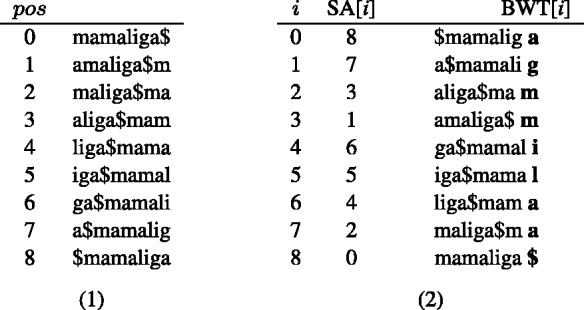
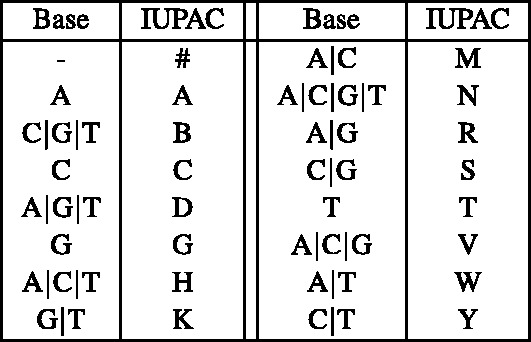
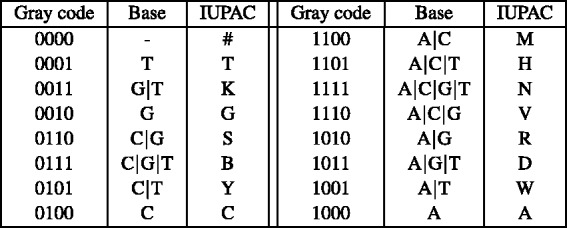
 nucleotide-to-bit assignment
nucleotide-to-bit assignment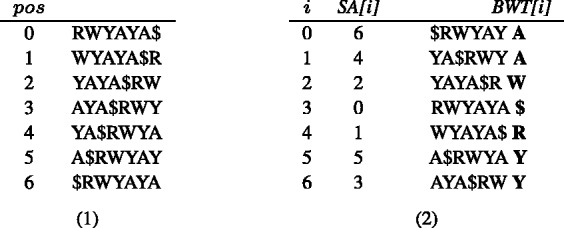
 G)(A
G)(A T)(C
T)(C T)A(C
T)A(C T)A). All the rotations of S are first listed (1) and then sorted in Gray code order (2)
T)A). All the rotations of S are first listed (1) and then sorted in Gray code order (2)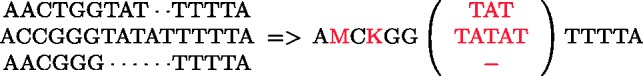



References
-
- Burrows M, Wheeler DJ. A block-sorting lossless data compression algorithm. Technical Report SRC-RR-124. 1994 HP Labs.
-
- Durbin M. So long, data depression. 2009. http://www.genomeweb.com/informatics/so-long-data-depression (31 May 2013, date last accessed)
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
