

Noncanonical matrix metalloprotease-1-protease-activated receptor-1 signaling triggers vascular smooth muscle cell dedifferentiation and arterial stenosis
- PMID: 23814055
- PMCID: PMC3743483
- DOI: 10.1074/jbc.M113.467019
Noncanonical matrix metalloprotease-1-protease-activated receptor-1 signaling triggers vascular smooth muscle cell dedifferentiation and arterial stenosis
Abstract
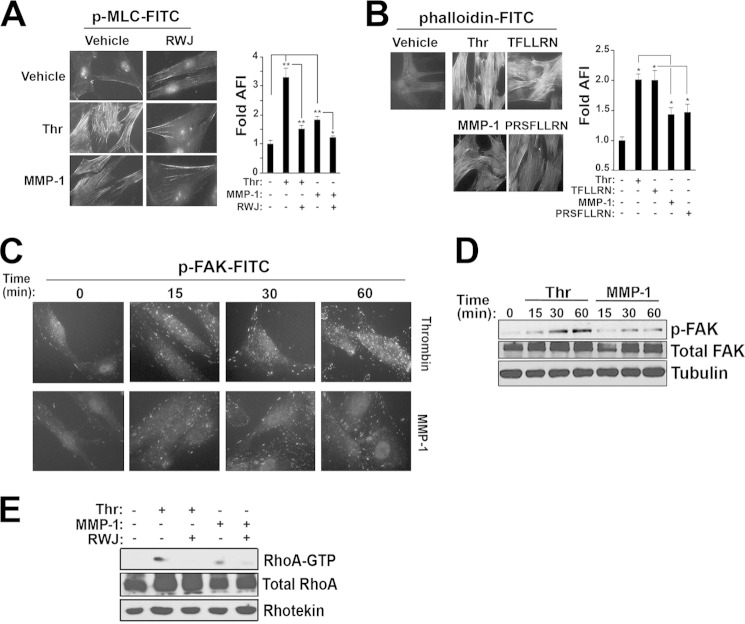
Vascular injury that results in proliferation and dedifferentiation of vascular smooth muscle cells (SMCs) is an important contributor to restenosis following percutaneous coronary interventions or plaque rupture. Protease-activated receptor-1 (PAR1) has been shown to play a role in vascular repair processes; however, little is known regarding its function or the relative roles of the upstream proteases thrombin and matrix metalloprotease-1 (MMP-1) in triggering PAR1-mediated arterial restenosis. The goal of this study was to determine whether noncanonical MMP-1 signaling through PAR1 would contribute to aberrant vascular repair processes in models of arterial injury. A mouse carotid arterial wire injury model was used for studies of neointima hyperplasia and arterial stenosis. The mice were treated post-injury for 21 days with a small molecule inhibitor of MMP-1 or a direct thrombin inhibitor and compared with vehicle control. Intimal and medial hyperplasia was significantly inhibited by 2.8-fold after daily treatment with the small molecule MMP-1 inhibitor, an effect that was lost in PAR1-deficient mice. Conversely, chronic inhibition of thrombin showed no benefit in suppressing the development of arterial stenosis. Thrombin-PAR1 signaling resulted in a supercontractile, differentiated phenotype in SMCs. Noncanonical MMP-1-PAR1 signaling resulted in the opposite effect and led to a dedifferentiated phenotype via a different G protein pathway. MMP-1-PAR1 significantly stimulated hyperplasia and migration of SMCs, and resulted in down-regulation of SMC contractile genes. These studies provide a new mechanism for the development of vascular intimal hyperplasia and suggest a novel therapeutic strategy to suppress restenosis by targeting noncanonical MMP-1-PAR1 signaling in vascular SMCs.
Keywords: Cardiovascular Disease; Cell Differentiation; Matrix Metalloproteinase (MMP); PAR1; Smooth Muscle; Thrombin.
Figures





References
-
- Heidenreich P. A., Trogdon J. G., Khavjou O. A., Butler J., Dracup K., Ezekowitz M. D., Finkelstein E. A., Hong Y., Johnston S. C., Khera A., Lloyd-Jones D. M., Nelson S. A., Nichol G., Orenstein D., Wilson P. W., Woo Y. J. (2011) Forecasting the future of cardiovascular disease in the United States. A policy statement from the American Heart Association. Circulation 123, 933–944 - PubMed
-
- Stone G. W., Maehara A., Lansky A. J., de Bruyne B., Cristea E., Mintz G. S., Mehran R., McPherson J., Farhat N., Marso S. P., Parise H., Templin B., White R., Zhang Z., Serruys P. W. (2011) A prospective natural-history study of coronary atherosclerosis. N. Engl. J. Med. 364, 226–235 - PubMed
-
- Owens G. K., Kumar M. S., Wamhoff B. R. (2004) Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol. Rev. 84, 767–801 - PubMed
-
- Smyth S. S., Pathak A., Stouffer G. A. (2002) In-stent restenosis. More fuel for the fire. Am. Heart J. 144, 577–579 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
