

Comparison of commercially available target enrichment methods for next-generation sequencing
- PMID: 23814499
- PMCID: PMC3605921
- DOI: 10.7171/jbt.13-2402-002
Comparison of commercially available target enrichment methods for next-generation sequencing
Abstract
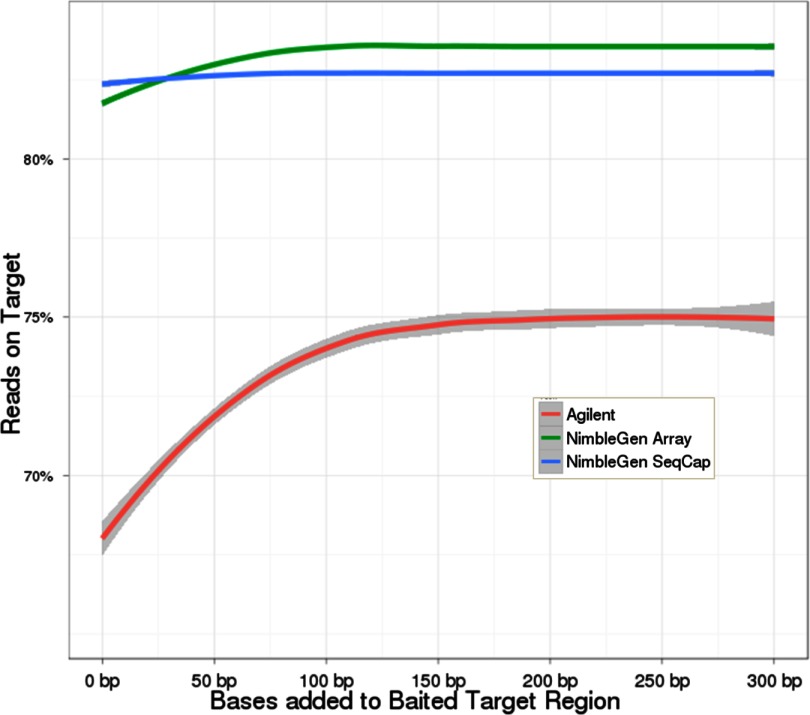
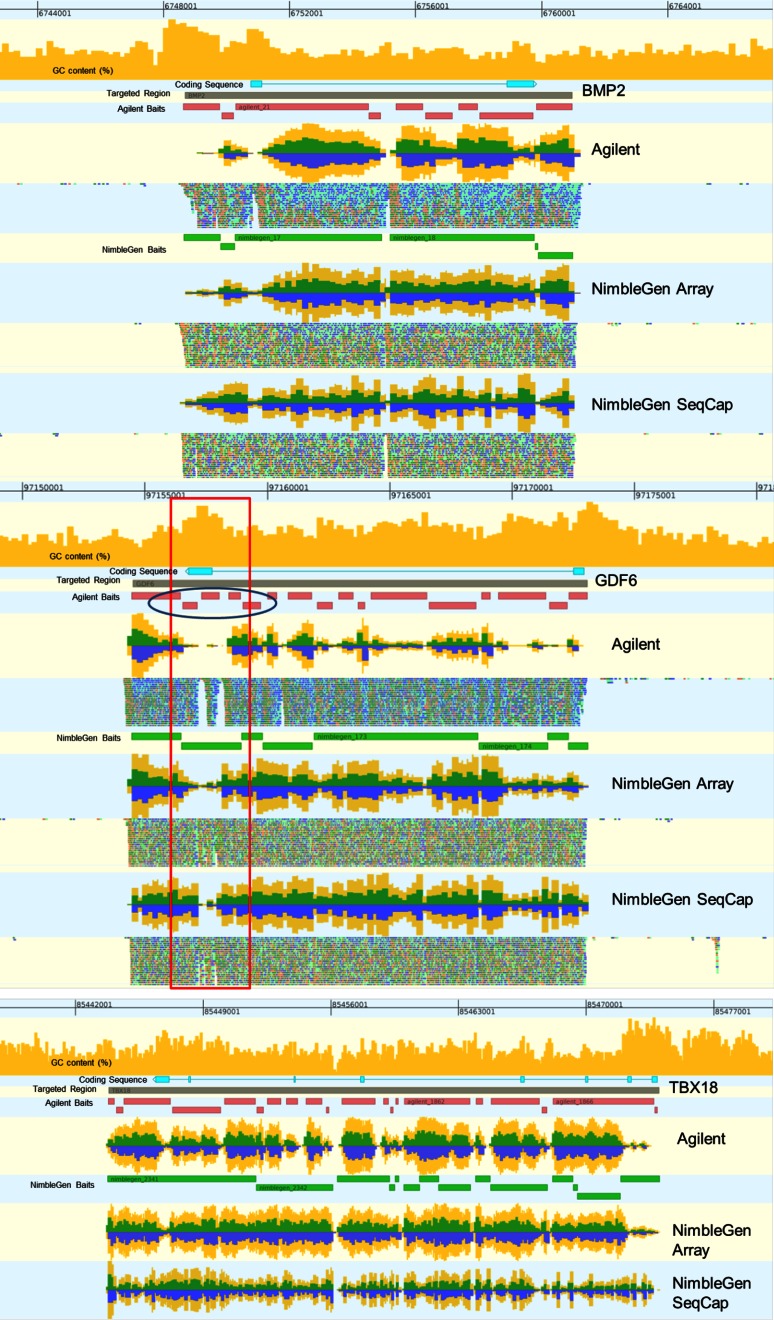
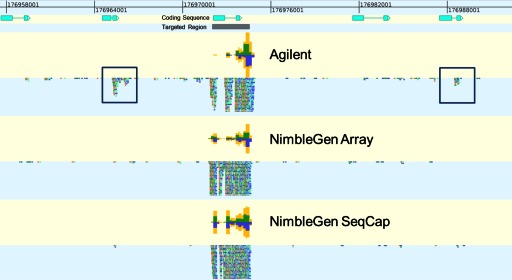
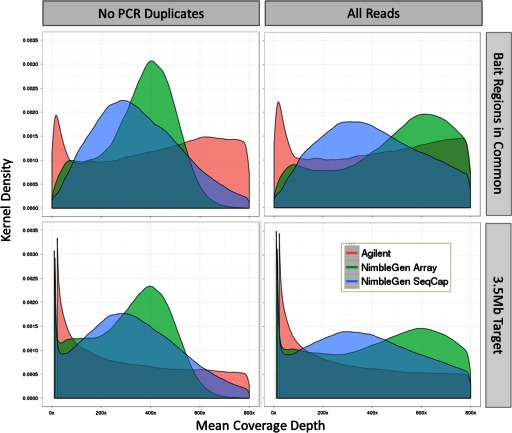
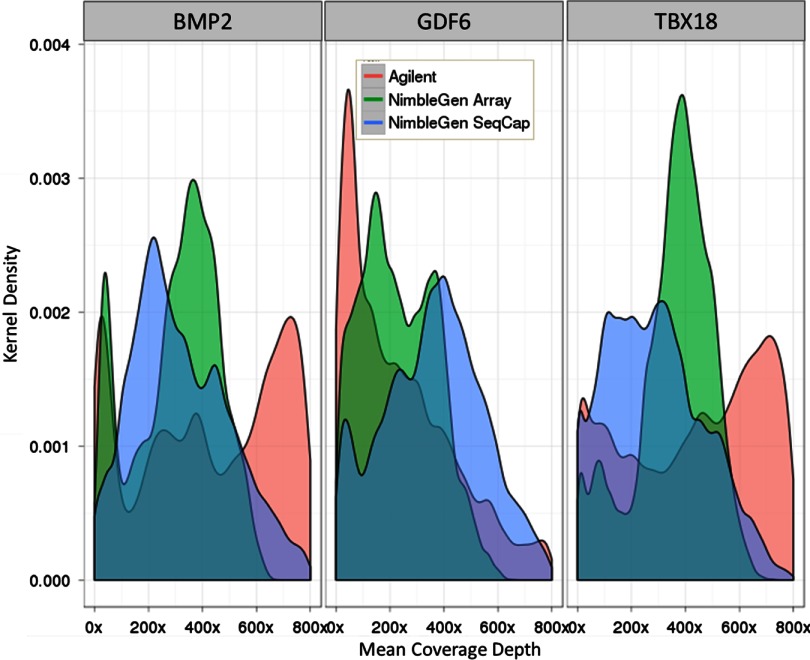
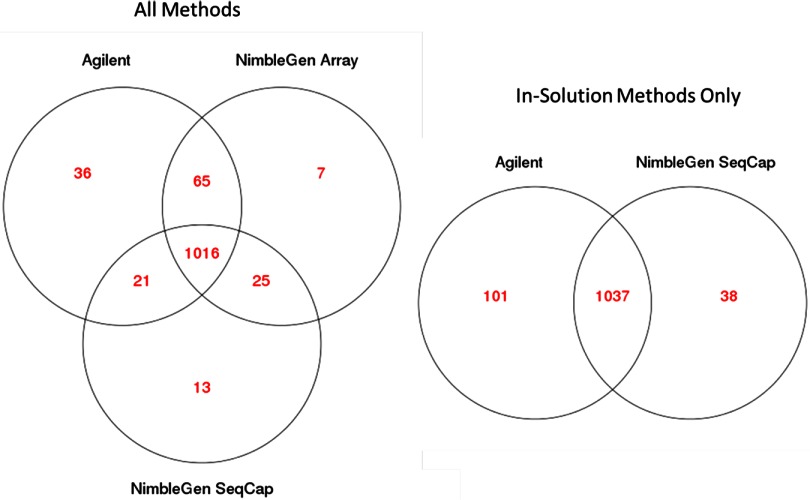
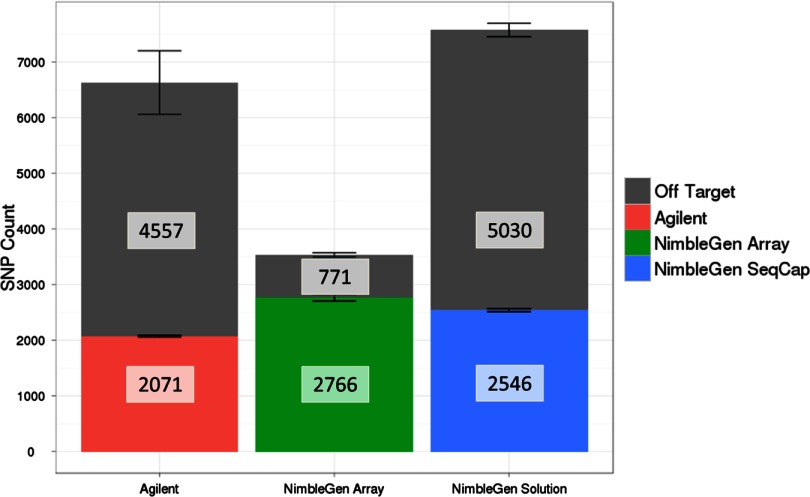
Isolating high-priority segments of genomes greatly enhances the efficiency of next-generation sequencing (NGS) by allowing researchers to focus on their regions of interest. For the 2010-11 DNA Sequencing Research Group (DSRG) study, we compared outcomes from two leading companies, Agilent Technologies (Santa Clara, CA, USA) and Roche NimbleGen (Madison, WI, USA), which offer custom-targeted genomic enrichment methods. Both companies were provided with the same genomic sample and challenged to capture identical genomic locations for DNA NGS. The target region totaled 3.5 Mb and included 31 individual genes and a 2-Mb contiguous interval. Each company was asked to design its best assay, perform the capture in replicates, and return the captured material to the DSRG-participating laboratories. Sequencing was performed in two different laboratories on Genome Analyzer IIx systems (Illumina, San Diego, CA, USA). Sequencing data were analyzed for sensitivity, specificity, and coverage of the desired regions. The success of the enrichment was highly dependent on the design of the capture probes. Overall, coverage variability was higher for the Agilent samples. As variant discovery is the ultimate goal for a typical targeted sequencing project, we compared samples for their ability to sequence single-nucleotide polymorphisms (SNPs) as a test of the ability to capture both chromosomes from the sample. In the targeted regions, we detected 2546 SNPs with the NimbleGen samples and 2071 with Agilent's. When limited to the regions that both companies included as baits, the number of SNPs was ∼1000 for each, with Agilent and NimbleGen finding a small number of unique SNPs not found by the other.
Keywords: Agilent; Illumina; NimbleGen; targeted capture.
Figures
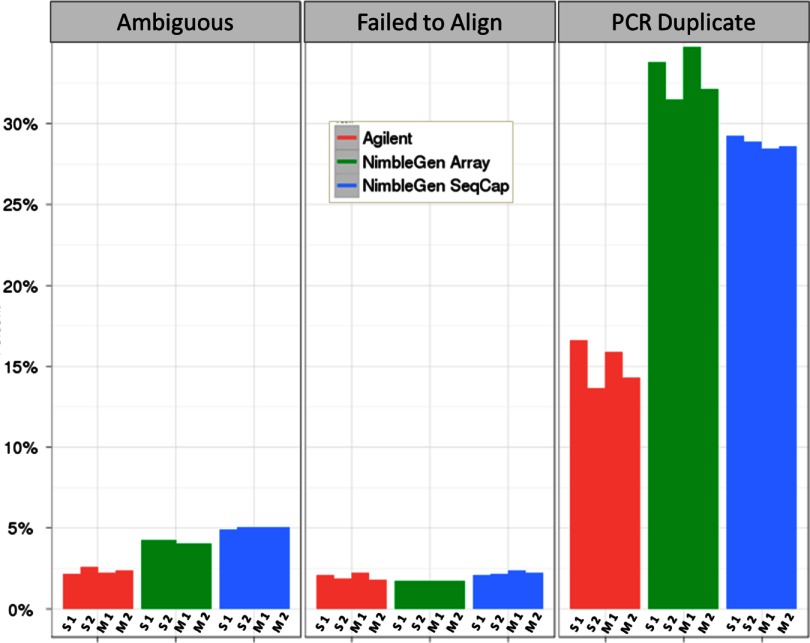
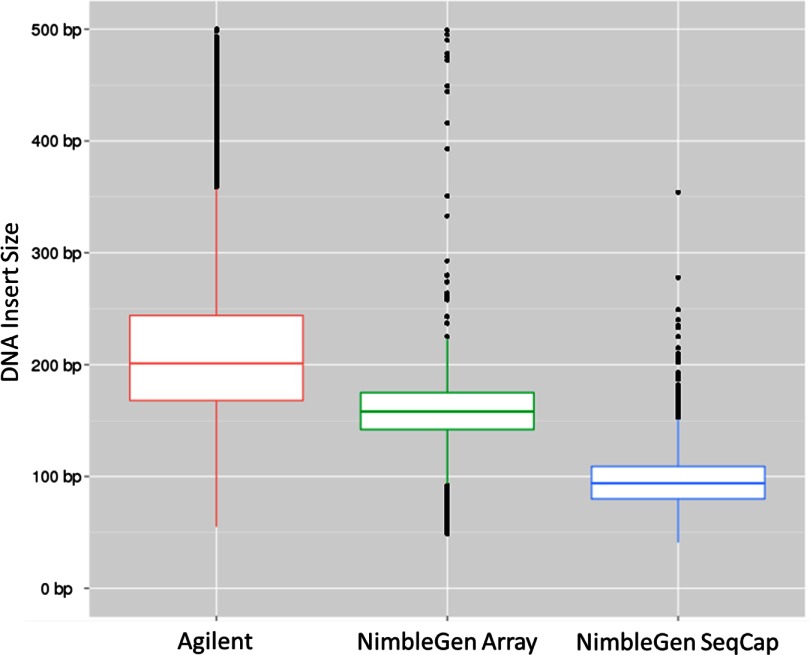
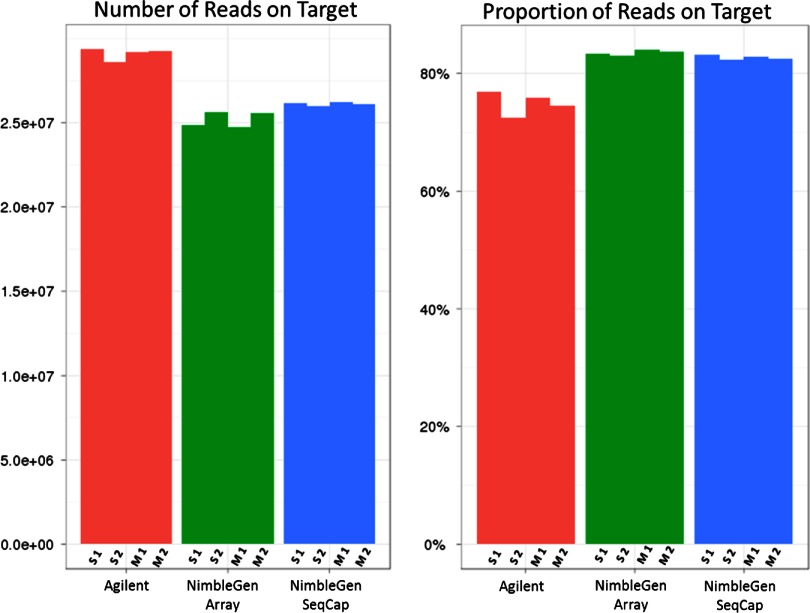
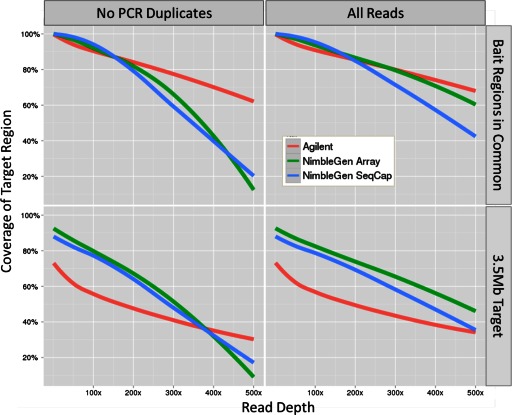
References
-
- Metzker ML. Sequencing technologies—the next generation. Nat Rev Genet 2010;11:31–46 - PubMed
-
- Tan IB, Cutcutache I, Zang ZJ, et al. Fanconi's anemia in adulthood: chemoradiation-induced bone marrow failure and a novel FANCA mutation identified by targeted deep sequencing. J Clin Oncol 2011;29:e591–e594 - PubMed
-
- Mamanova L, Coffey AJ, Scott CE, et al. Target-enrichment strategies for next-generation sequencing. Nat Methods 2010;7:111–118 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous