

Kraken: a set of tools for quality control and analysis of high-throughput sequence data
- PMID: 23816787
- PMCID: PMC3991327
- DOI: 10.1016/j.ymeth.2013.06.027
Kraken: a set of tools for quality control and analysis of high-throughput sequence data
Abstract
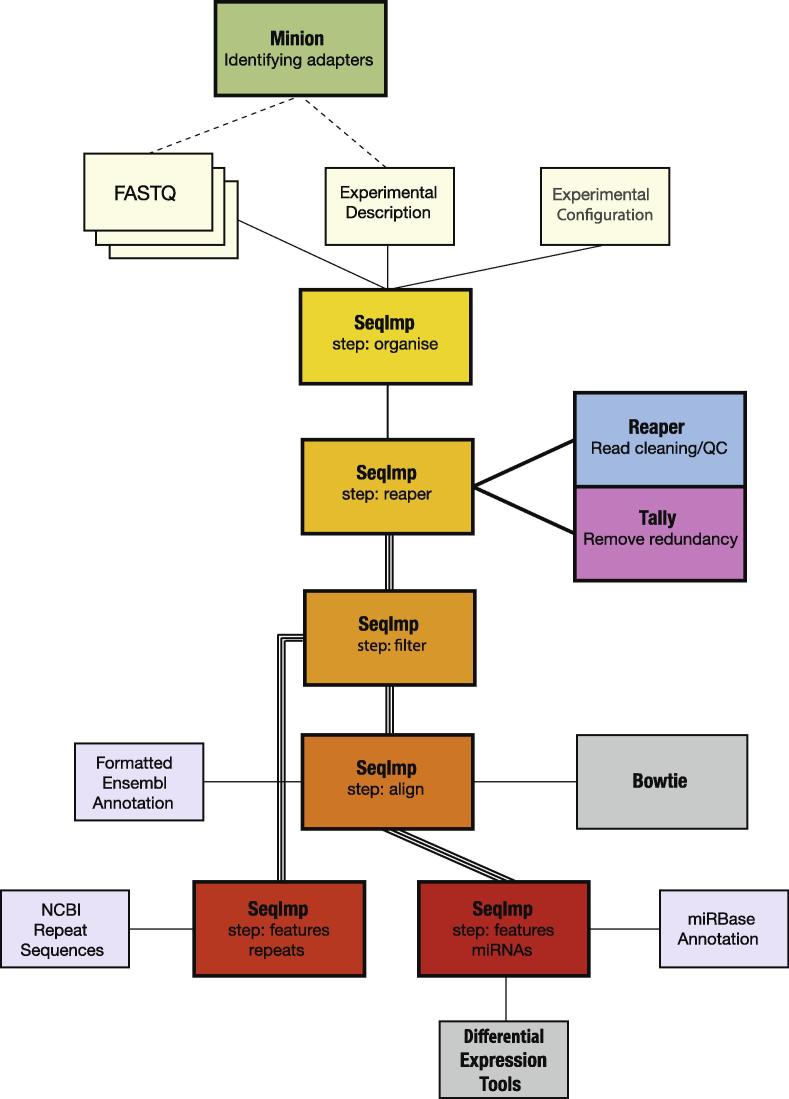
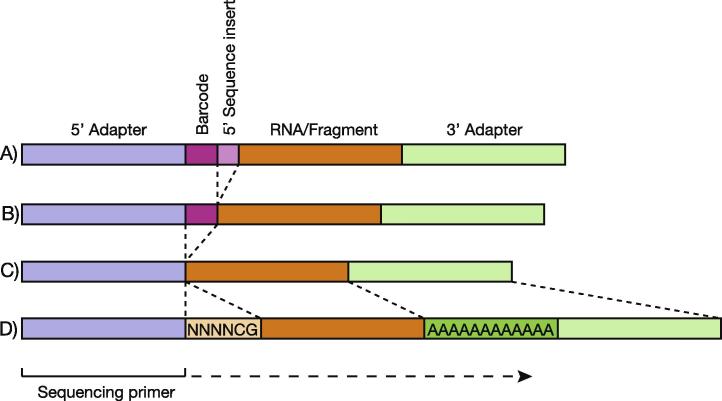
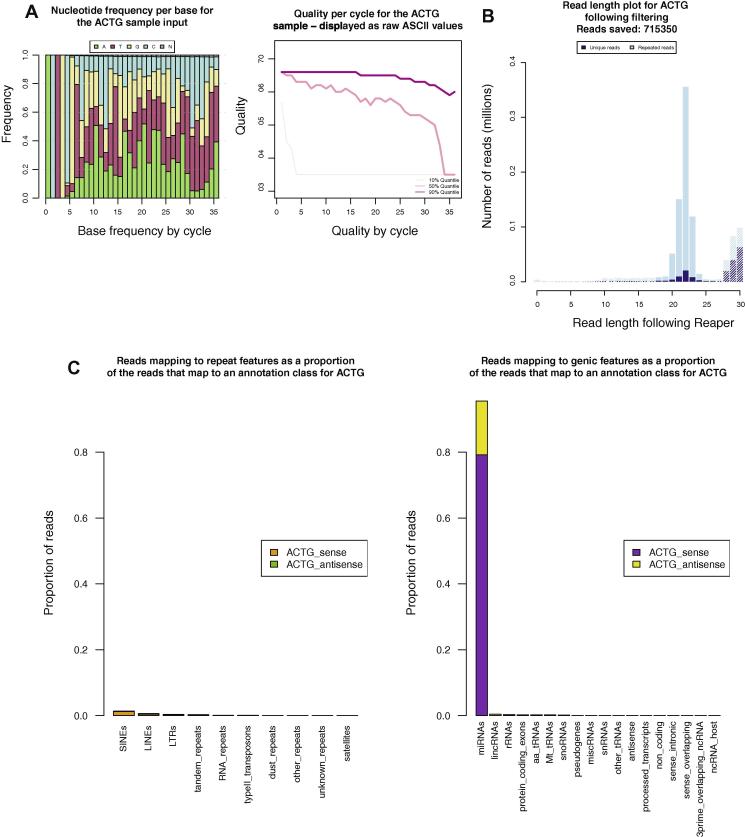
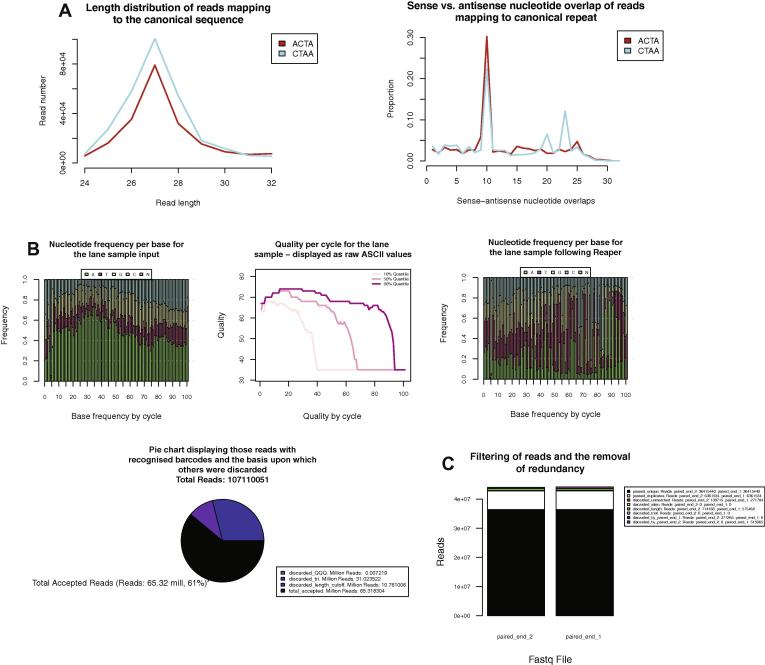
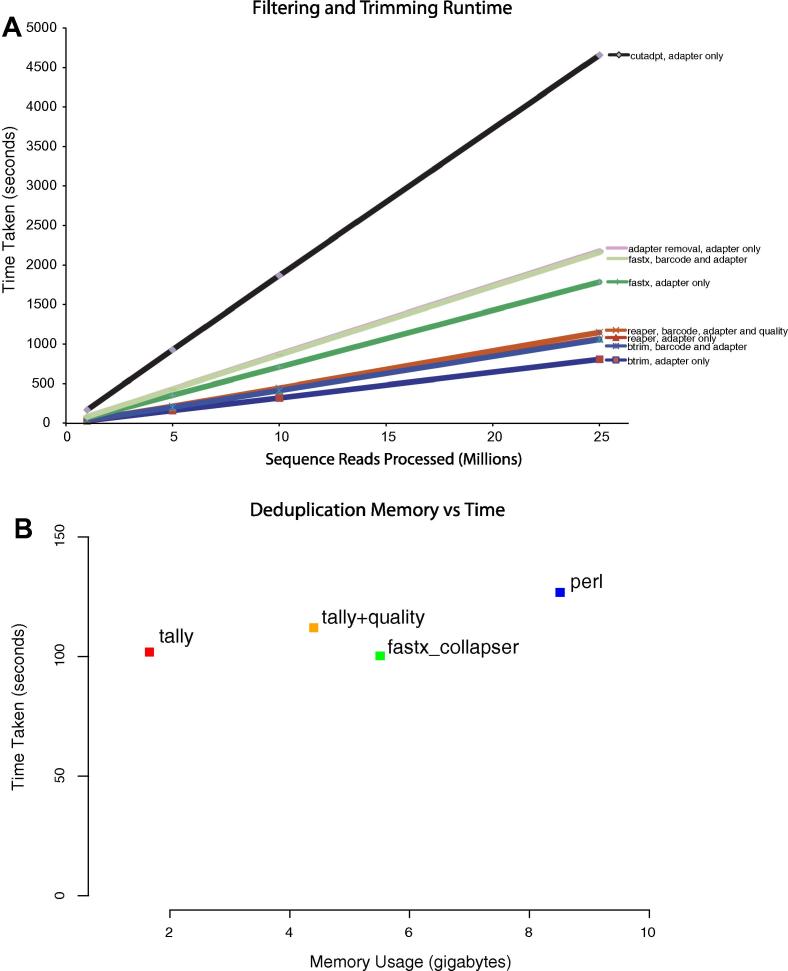
New sequencing technologies pose significant challenges in terms of data complexity and magnitude. It is essential that efficient software is developed with performance that scales with this growth in sequence information. Here we present a comprehensive and integrated set of tools for the analysis of data from large scale sequencing experiments. It supports adapter detection and removal, demultiplexing of barcodes, paired-end data, a range of read architectures and the efficient removal of sequence redundancy. Sequences can be trimmed and filtered based on length, quality and complexity. Quality control plots track sequence length, composition and summary statistics with respect to genomic annotation. Several use cases have been integrated into a single streamlined pipeline, including both mRNA and small RNA sequencing experiments. This pipeline interfaces with existing tools for genomic mapping and differential expression analysis.
Keywords: Adapter trimming; Algorithms; NGS; Next-generation sequencing; Pipelines; RNAseq; Sequencing; Tools.
Copyright © 2013 The Authors. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Gunaratne P.H., Coarfa C., Soibam B., Tandon A. Methods Mol Biol (Clifton, NJ) 2012;822:273–288. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
