

Standard care versus protocol based therapy for new onset Pseudomonas aeruginosa in cystic fibrosis
- PMID: 23818295
- PMCID: PMC4059359
- DOI: 10.1002/ppul.22693
Standard care versus protocol based therapy for new onset Pseudomonas aeruginosa in cystic fibrosis
Abstract
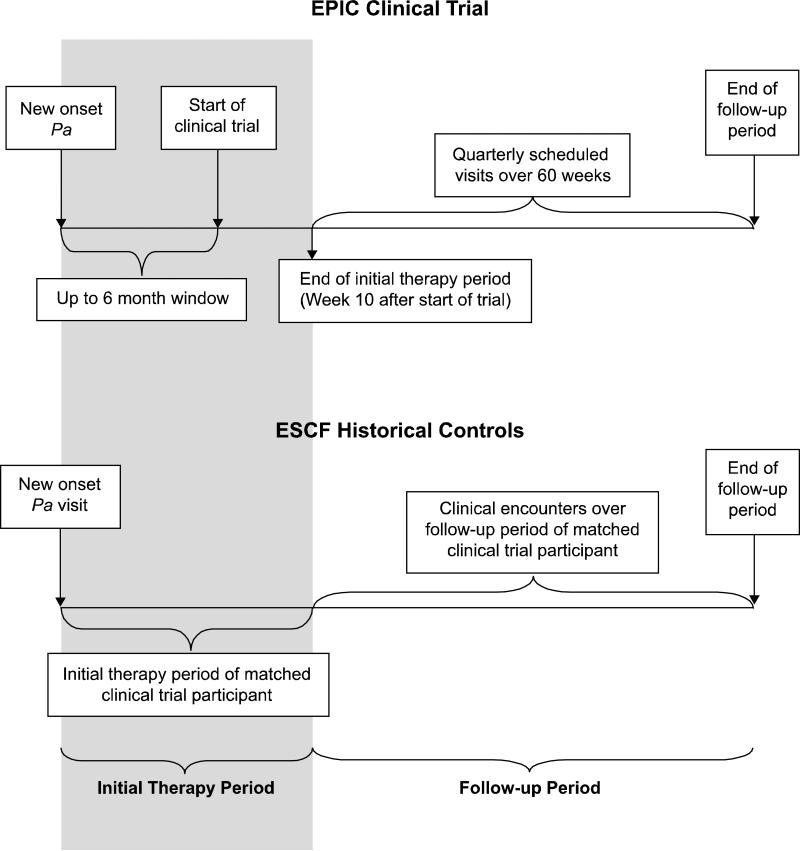
Rationale: The Early Pseudomonal Infection Control (EPIC) randomized trial rigorously evaluated the efficacy of different antibiotic regimens for eradication of newly identified Pseudomonas (Pa) in children with cystic fibrosis (CF). Protocol based therapy in the trial was provided based on culture positivity independent of symptoms. It is unclear whether outcomes observed in the clinical trial were different than those that would have been observed with historical standard of care driven more heavily by respiratory symptoms than culture positivity alone. We hypothesized that the incidence of Pa recurrence and hospitalizations would be significantly reduced among trial participants as compared to historical controls whose standard of care preceded the widespread adoption of tobramycin inhalation solution (TIS) as initial eradication therapy at the time of new isolation of Pa.
Methods: Eligibility criteria from the trial were used to derive historical controls from the Epidemiologic Study of CF (ESCF) who received standard of care treatment from 1995 to 1998, before widespread availability of TIS. Pa recurrence and hospitalization outcomes were assessed over a 15-month time period.
Results: As compared to 100% of the 304 trial participants, only 296/608 (49%) historical controls received antibiotics within an average of 20 weeks after new onset Pa. Pa recurrence occurred among 104/298 (35%) of the trial participants as compared to 295/549 (54%) of historical controls (19% difference, 95% CI: 12%, 26%, P < 0.001). No significant differences in the incidence of hospitalization were observed between cohorts.
Conclusions: Protocol-based antimicrobial therapy for newly acquired Pa resulted in a lower rate of Pa recurrence but comparable hospitalization rates as compared to a historical control cohort less aggressively treated with antibiotics for new onset Pa.
Keywords: Pseudomonas aeruginosa; cystic fibrosis; early intervention; historical controls; randomized trial.
© 2013 Wiley Periodicals, Inc.
Figures
References
-
- Welsh MJRB, Accurso F. Cystic Fibrosis. In: Scriver CRBAL, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. 8th ed. McGraw-Hill; New York, NY: 2001. pp. 521–88.
-
- Gibson RL, Burns JL, Ramsey BW. Pathophysiology and management of pulmonary infections in cystic fibrosis. Am J Respir Crit Care Med. 2003;168(8):918–51. - PubMed
-
- Pamukcu A, Bush A, Buchdahl R. Effects of Pseudomonas aeruginosa colonization on lung function and anthropometric variables in children with cystic fibrosis. Pediatr Pulmonol. 1995;19(1):10–5. - PubMed
-
- Henry RL, Mellis CM, Petrovic L. Mucoid Pseudomonas aeruginosa is a marker of poor survival in cystic fibrosis. Pediatr Pulmonol. 1992;12(3):158–61. - PubMed
-
- Kosorok MR, Zeng L, West SE, Rock MJ, Splaingard ML, Laxova A, Green CG, Collins J, Farrell PM. Acceleration of lung disease in children with cystic fibrosis after Pseudomonas aeruginosa acquisition. Pediatr Pulmonol. 2001;32(4):277–87. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
