

Assessment of computational methods for predicting the effects of missense mutations in human cancers
- PMID: 23819521
- PMCID: PMC3665581
- DOI: 10.1186/1471-2164-14-S3-S7
Assessment of computational methods for predicting the effects of missense mutations in human cancers
Abstract
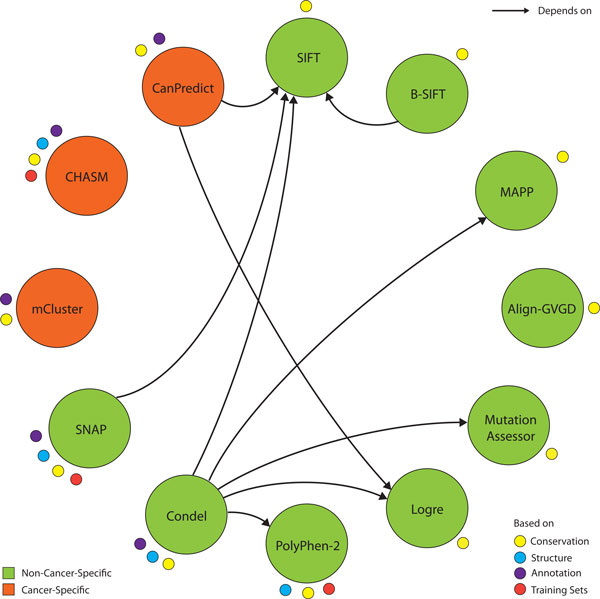
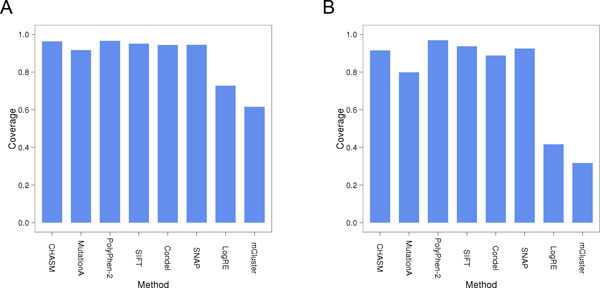
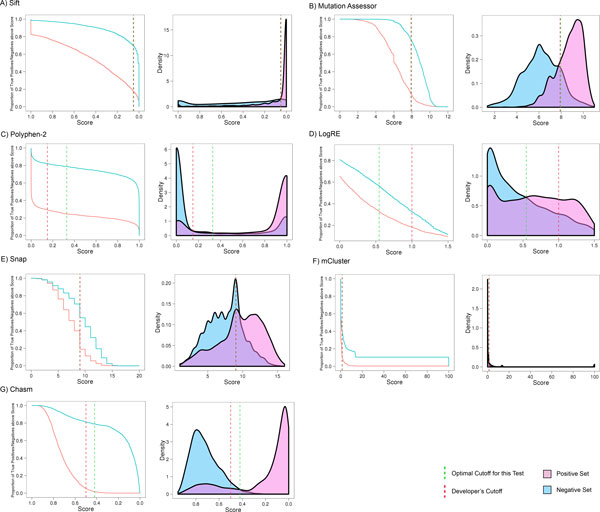
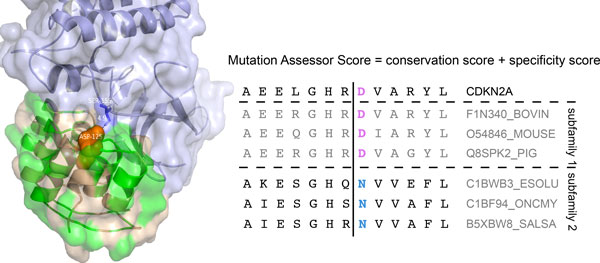
Background: Recent advances in sequencing technologies have greatly increased the identification of mutations in cancer genomes. However, it remains a significant challenge to identify cancer-driving mutations, since most observed missense changes are neutral passenger mutations. Various computational methods have been developed to predict the effects of amino acid substitutions on protein function and classify mutations as deleterious or benign. These include approaches that rely on evolutionary conservation, structural constraints, or physicochemical attributes of amino acid substitutions. Here we review existing methods and further examine eight tools: SIFT, PolyPhen2, Condel, CHASM, mCluster, logRE, SNAP, and MutationAssessor, with respect to their coverage, accuracy, availability and dependence on other tools.
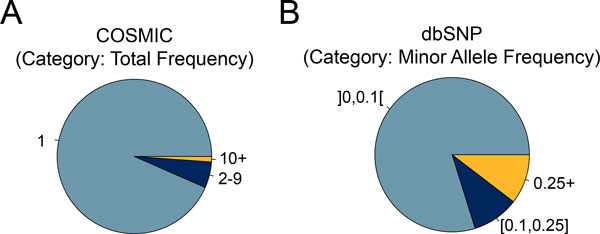
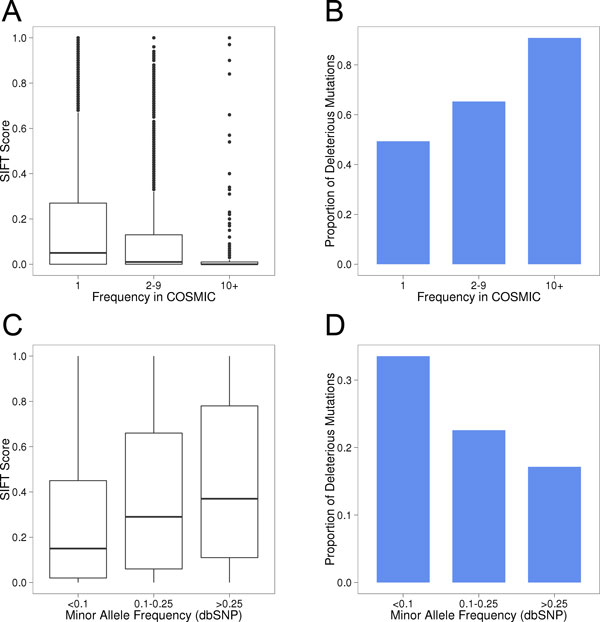
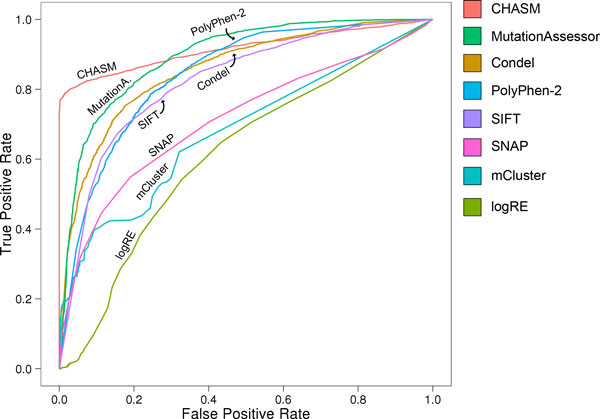
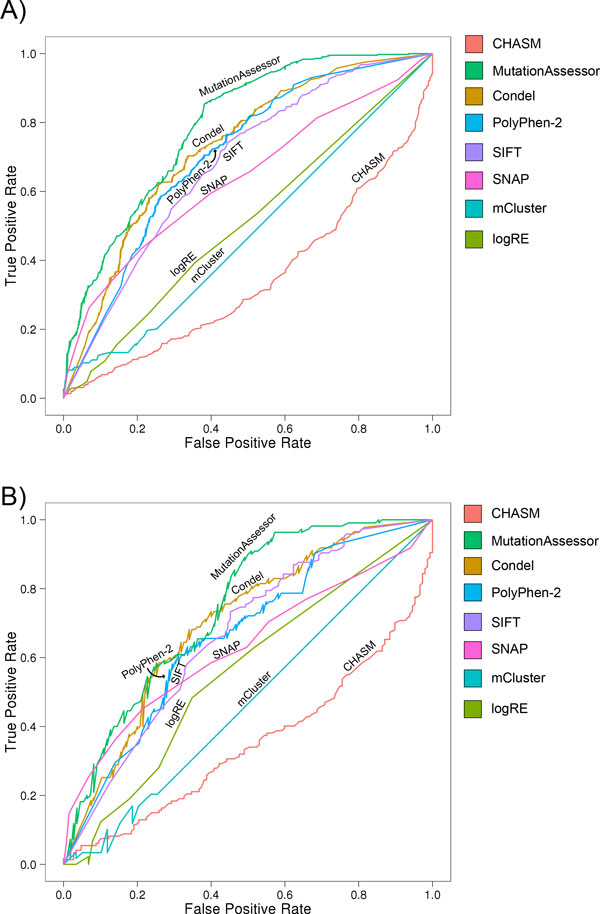
Results: Single nucleotide polymorphisms with high minor allele frequencies were used as a negative (neutral) set for testing, and recurrent mutations from the COSMIC database as well as novel recurrent somatic mutations identified in very recent cancer studies were used as positive (non-neutral) sets. Conservation-based methods generally had moderately high accuracy in distinguishing neutral from deleterious mutations, whereas the performance of machine learning based predictors with comprehensive feature spaces varied between assessments using different positive sets. MutationAssessor consistently provided the highest accuracies. For certain combinations metapredictors slightly improved the performance of included individual methods, but did not outperform MutationAssessor as stand-alone tool.
Conclusions: Our independent assessment of existing tools reveals various performance disparities. Cancer-trained methods did not improve upon more general predictors. No method or combination of methods exceeds 81% accuracy, indicating there is still significant room for improvement for driver mutation prediction, and perhaps more sophisticated feature integration is needed to develop a more robust tool.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
