

Niemann-pick disease type C: new aspects in a long published family - partial manifestations in heterozygotes
- PMID: 23821321
- PMCID: PMC3897806
- DOI: 10.1007/8904_2013_240
Niemann-pick disease type C: new aspects in a long published family - partial manifestations in heterozygotes
Abstract
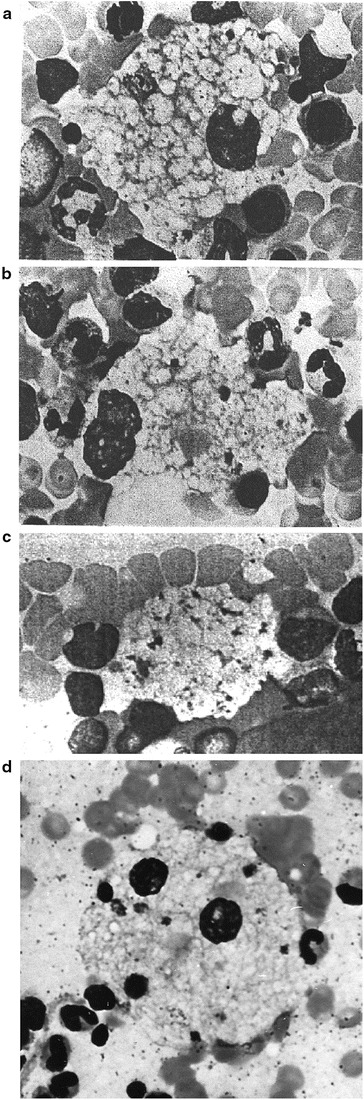
Decades ago, a family with three children with a neurovisceral lysosomal storage disease was described. The patient siblings died at ages 7, 9, and 11 years, respectively, and according to the current concept had the late-infantile neurologic form of Niemann-Pick type C1 (NPC) disease, given by the present molecular study that there were severe NPC1 gene variants: Blood samples preserved since that time from one patient sibling and his presently 55-year-old essentially healthy sister have now been studied, revealing the variants p.I1061T and p.G1162V in the NPC1 gene, the first long known, the second newly found but predicted to be pathogenic and similar to the known G1162A. Now, with the molecular diagnosis, that initial description warrants new interest for the following reasons. The mentioned sister carries only the I1061T variant. She had storage macrophages ("Niemann-Pick cells") in her bone marrow, but also displayed distinct splenomegaly with indurated consistency of the organ, proven in childhood and confirmed several times up to age 13, but disappeared at age 55 years. She shares the I1061T variant with her still healthy mother, and the bone marrow finding with both parents, her father having died at 66 years from a carcinoma. The present study is one of the first describing hematological and relevant clinical symptomatology, even in heterozygotes, of molecularly diagnosed human NPC. Feline NPC is known to model such a situation. For human diagnostic and clinical NPC management, the possibility of "heterozygous disease" should be kept in mind.
Figures

Similar articles
-
Phenotypic variability of Niemann-Pick disease type C including a case with clinically pure schizophrenia: a case report.BMC Neurol. 2018 Aug 17;18(1):117. doi: 10.1186/s12883-018-1124-2. BMC Neurol. 2018. PMID: 30119649 Free PMC article.
-
[Niemann-Pick type C disease and psychosis: Two siblings].Encephale. 2015 Jun;41(3):238-43. doi: 10.1016/j.encep.2014.08.007. Epub 2014 Sep 18. Encephale. 2015. PMID: 25238906 French.
-
Severe demyelination in a patient with a late infantile form of Niemann-Pick disease type C.Neuropathology. 2017 Oct;37(5):426-430. doi: 10.1111/neup.12380. Epub 2017 Apr 7. Neuropathology. 2017. PMID: 28387450
-
[Adult onset Niemann-Pick type C disease and psychosis: literature review].Encephale. 2013 Oct;39(5):315-9. doi: 10.1016/j.encep.2013.04.013. Epub 2013 Aug 5. Encephale. 2013. PMID: 23928063 Review. French.
-
Lipid changes in Niemann-Pick disease type C brain: personal experience and review of the literature.Neurochem Res. 1999 Apr;24(4):481-9. doi: 10.1023/a:1022575511354. Neurochem Res. 1999. PMID: 10227680 Review.
Cited by
-
The Extending Spectrum of NPC1-Related Human Disorders: From Niemann-Pick C1 Disease to Obesity.Endocr Rev. 2018 Apr 1;39(2):192-220. doi: 10.1210/er.2017-00176. Endocr Rev. 2018. PMID: 29325023 Free PMC article. Review.
-
Current concepts in the neuropathogenesis of mucolipidosis type IV.J Neurochem. 2019 Mar;148(5):669-689. doi: 10.1111/jnc.14462. Epub 2018 Aug 30. J Neurochem. 2019. PMID: 29770442 Free PMC article. Review.
-
Pulmonary Involvement in Niemann-Pick Disease: A State-of-the-Art Review.Lung. 2016 Aug;194(4):511-8. doi: 10.1007/s00408-016-9893-0. Epub 2016 May 10. Lung. 2016. PMID: 27164983 Review.
-
Phenotypic variability of Niemann-Pick disease type C including a case with clinically pure schizophrenia: a case report.BMC Neurol. 2018 Aug 17;18(1):117. doi: 10.1186/s12883-018-1124-2. BMC Neurol. 2018. PMID: 30119649 Free PMC article.
-
Different Niemann-Pick C1 Genotypes Generate Protein Phenotypes that Vary in their Intracellular Processing, Trafficking and Localization.Sci Rep. 2019 Mar 28;9(1):5292. doi: 10.1038/s41598-019-41707-y. Sci Rep. 2019. PMID: 30923329 Free PMC article.
References
LinkOut - more resources
Full Text Sources
Other Literature Sources
