

Mutation rates, spectra, and genome-wide distribution of spontaneous mutations in mismatch repair deficient yeast
- PMID: 23821616
- PMCID: PMC3755907
- DOI: 10.1534/g3.113.006429
Mutation rates, spectra, and genome-wide distribution of spontaneous mutations in mismatch repair deficient yeast
Abstract
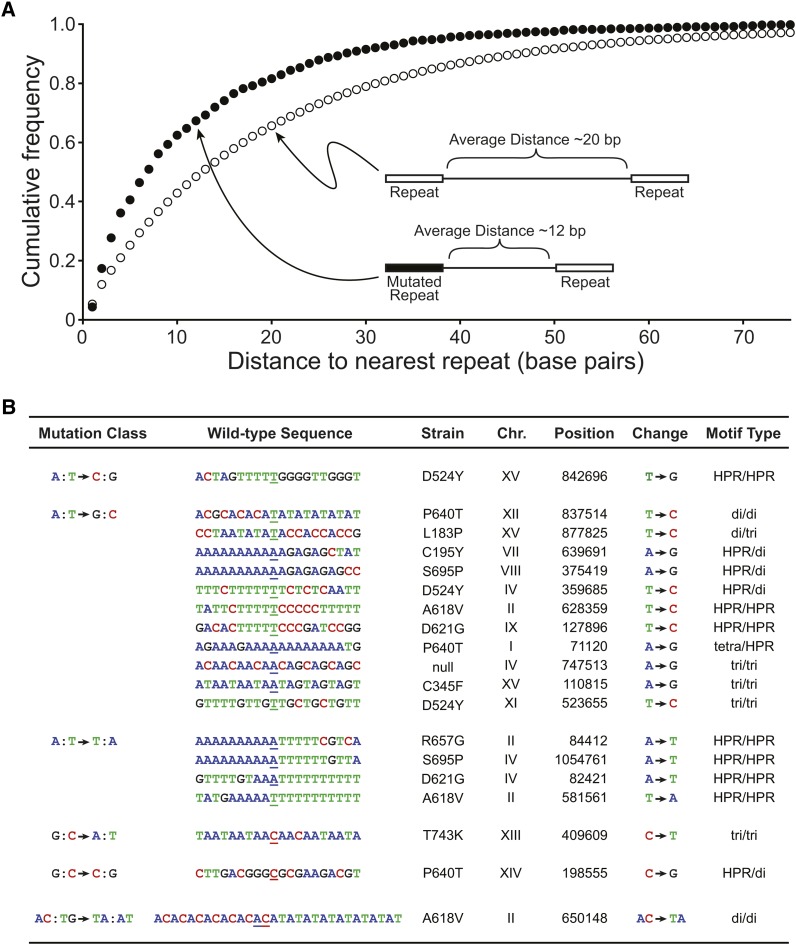
DNA mismatch repair is a highly conserved DNA repair pathway. In humans, germline mutations in hMSH2 or hMLH1, key components of mismatch repair, have been associated with Lynch syndrome, a leading cause of inherited cancer mortality. Current estimates of the mutation rate and the mutational spectra in mismatch repair defective cells are primarily limited to a small number of individual reporter loci. Here we use the yeast Saccharomyces cerevisiae to generate a genome-wide view of the rates, spectra, and distribution of mutation in the absence of mismatch repair. We performed mutation accumulation assays and next generation sequencing on 19 strains, including 16 msh2 missense variants implicated in Lynch cancer syndrome. The mutation rate for DNA mismatch repair null strains was approximately 1 mutation per genome per generation, 225-fold greater than the wild-type rate. The mutations were distributed randomly throughout the genome, independent of replication timing. The mutation spectra included insertions/deletions at homopolymeric runs (87.7%) and at larger microsatellites (5.9%), as well as transitions (4.5%) and transversions (1.9%). Additionally, repeat regions with proximal repeats are more likely to be mutated. A bias toward deletions at homopolymers and insertions at (AT)n microsatellites suggests a different mechanism for mismatch generation at these sites. Interestingly, 5% of the single base pair substitutions might represent double-slippage events that occurred at the junction of immediately adjacent repeats, resulting in a shift in the repeat boundary. These data suggest a closer scrutiny of tumor suppressors with homopolymeric runs with proximal repeats as the potential drivers of oncogenesis in mismatch repair defective cells.
Keywords: homopolymeric runs; microsatellites; mismatch repair; mutation accumulation; mutation rate.
Figures



References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases