

Involvement of exercise-induced macrophage migration inhibitory factor in the prevention of fatty liver disease
- PMID: 23823021
- PMCID: PMC3757527
- DOI: 10.1530/JOE-13-0135
Involvement of exercise-induced macrophage migration inhibitory factor in the prevention of fatty liver disease
Abstract
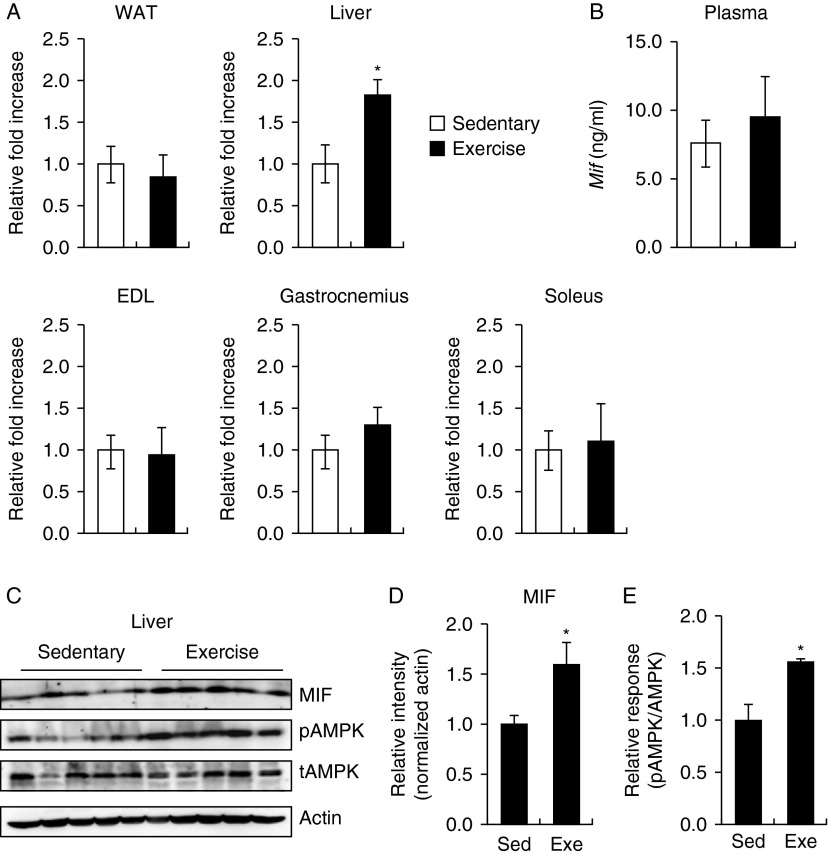
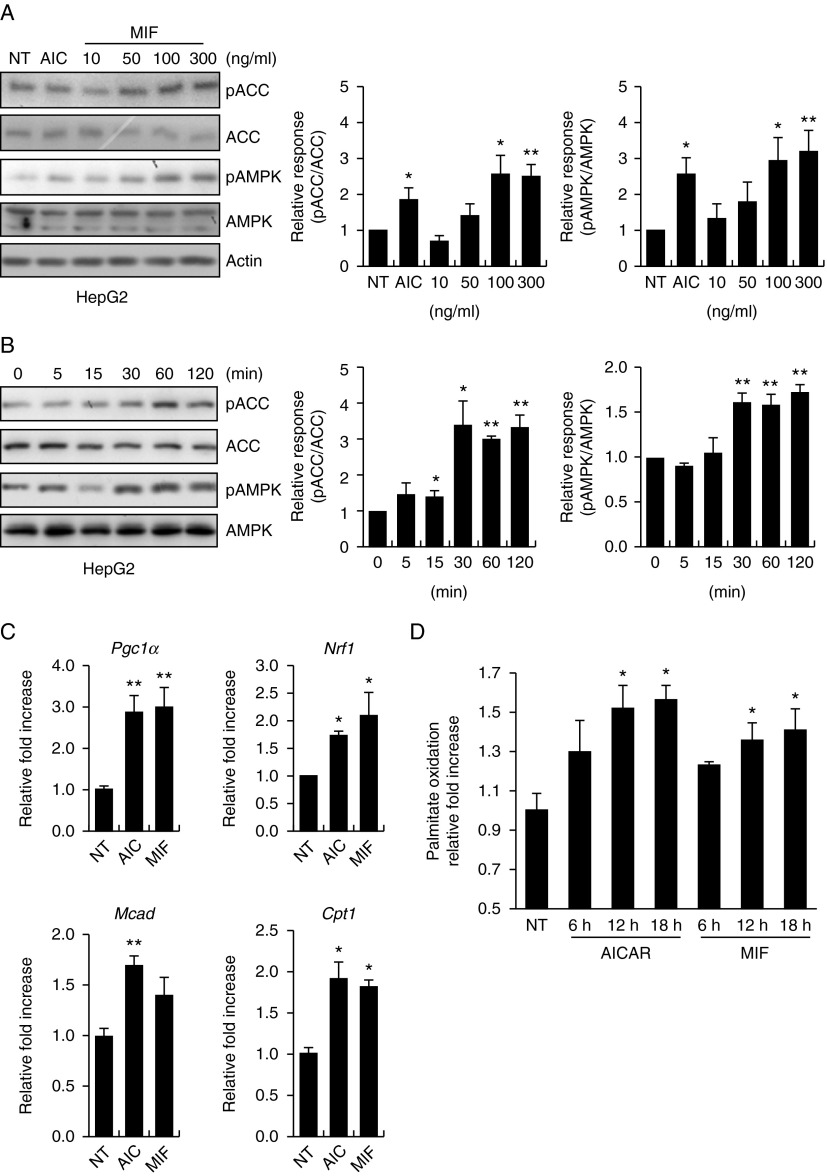
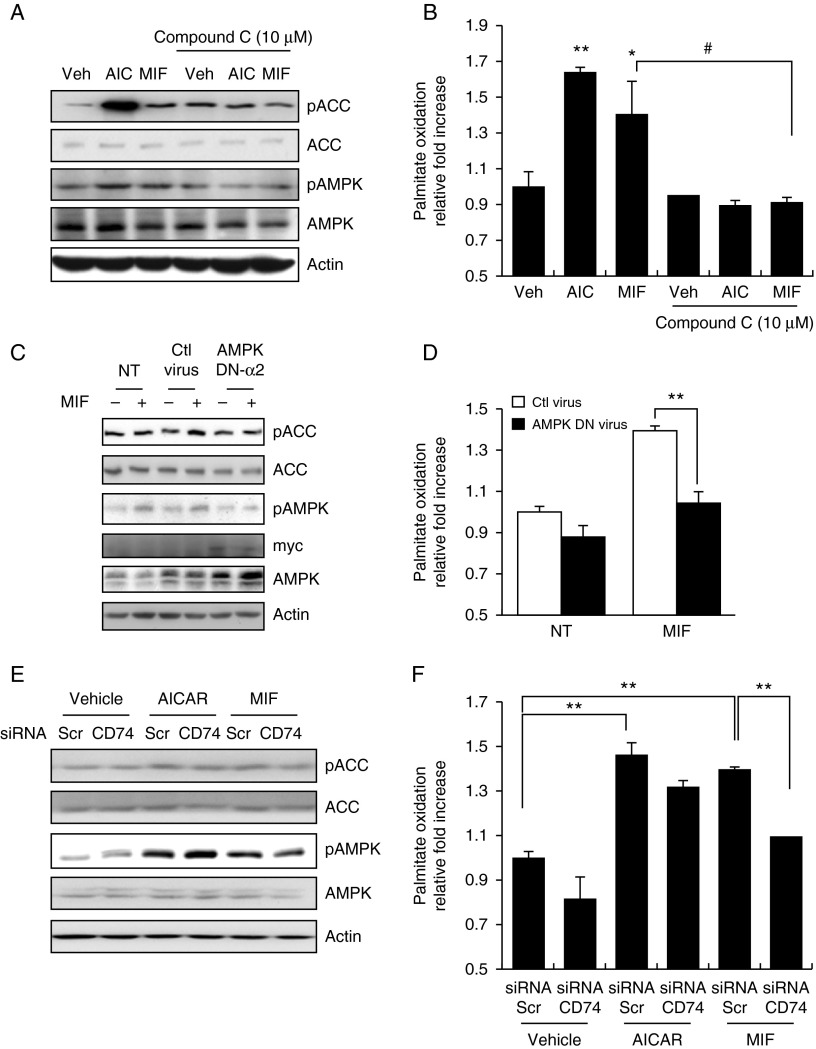
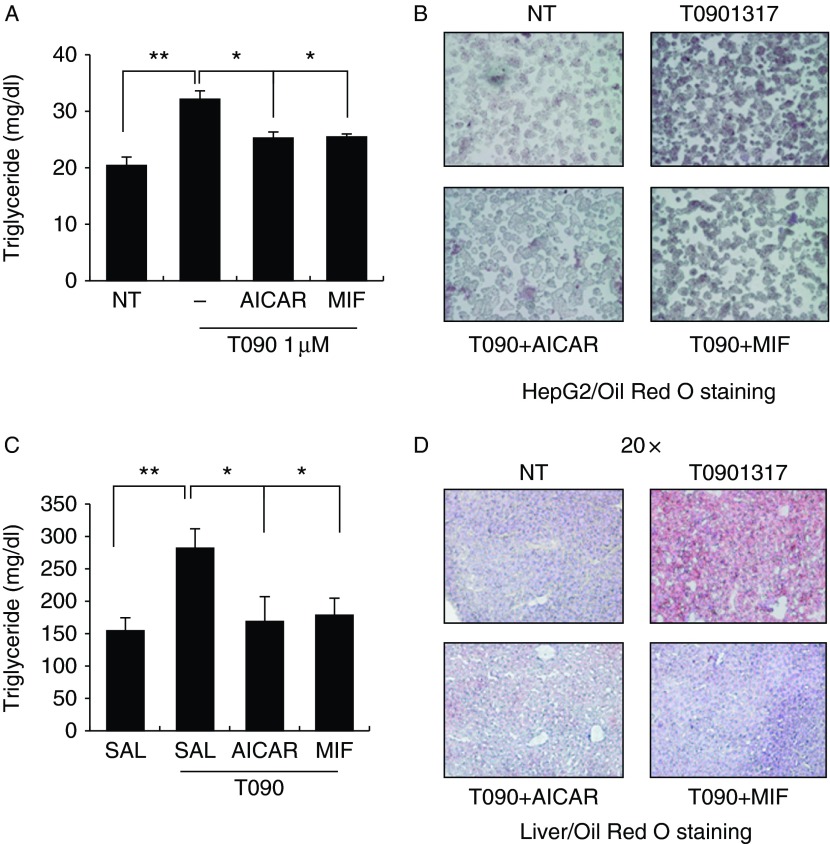
Physical inactivity can lead to obesity and fat accumulation in various tissues. Critical complications of obesity include type II diabetes and nonalcoholic fatty liver disease (NAFLD). Exercise has been reported to have ameliorating effects on obesity and NAFLD. However, the underlying mechanism is not fully understood. We showed that liver expression of macrophage migration inhibitory factor (MIF) was increased after 4 weeks of treadmill exercise. Phosphorylation of AMP-activated protein kinase and acetyl-CoA carboxylase in human hepatocyte cell lines was enhanced after MIF treatment. These responses were accompanied by increases in lipid oxidation. Moreover, inhibition of either AMPK or cluster of differentiation 74 resulted in inhibition of MIF-induced lipid oxidation. Furthermore, the administration of MIF to a human hepatocyte cell line and mice liver reduced liver X receptor agonist-induced lipid accumulation. Taken together, these results indicate that MIF is highly expressed in the liver during physical exercise and may prevent hepatic steatosis by activating the AMPK pathway.
Keywords: cytokines; exercise; lipid; liver; molecular biology.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
