

Determinants of myocardial conduction velocity: implications for arrhythmogenesis
- PMID: 23825462
- PMCID: PMC3695374
- DOI: 10.3389/fphys.2013.00154
Determinants of myocardial conduction velocity: implications for arrhythmogenesis
Abstract
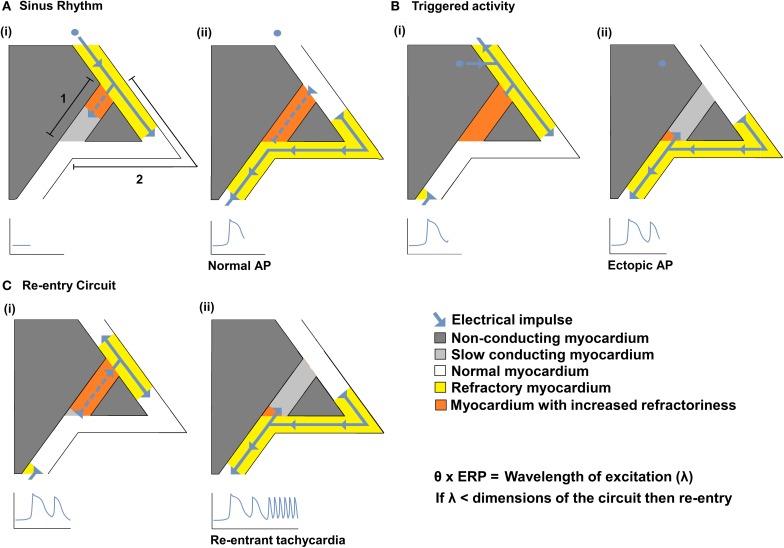
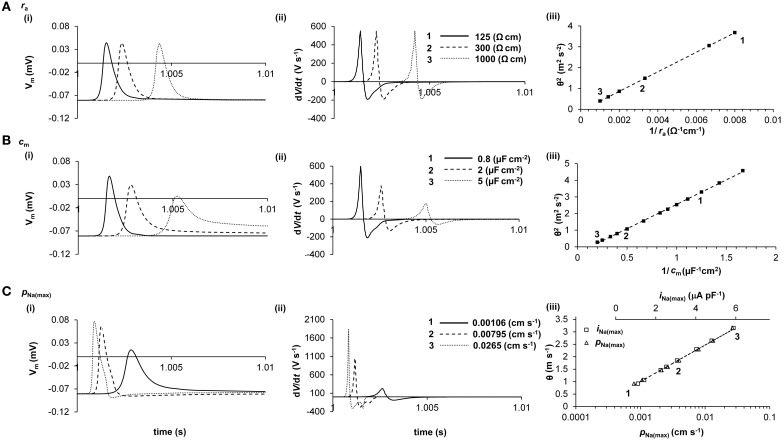
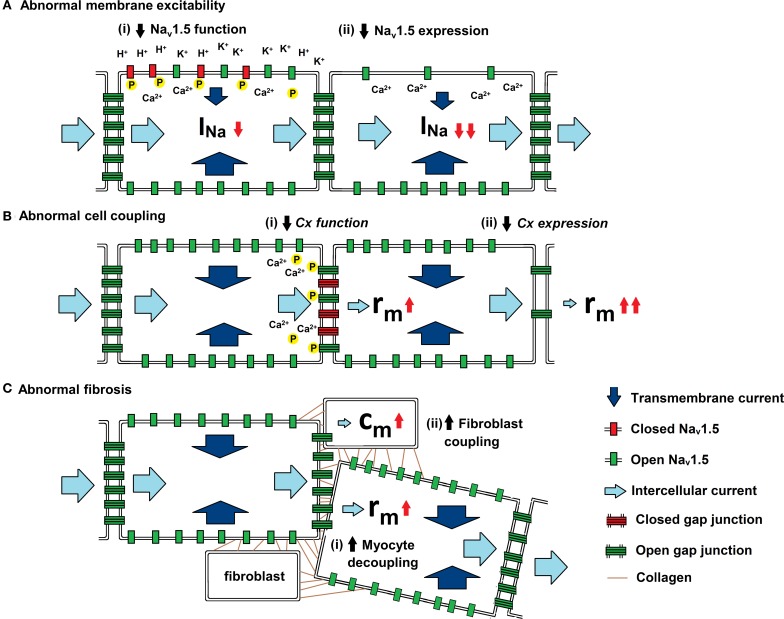
Slowed myocardial conduction velocity (θ) is associated with an increased risk of re-entrant excitation, predisposing to cardiac arrhythmia. θ is determined by the ion channel and physical properties of cardiac myocytes and by their interconnections. Thus, θ is closely related to the maximum rate of action potential (AP) depolarization [(dV/dt)max], as determined by the fast Na(+) current (I Na); the axial resistance (r a) to local circuit current flow between cells; their membrane capacitances (c m); and to the geometrical relationship between successive myocytes within cardiac tissue. These determinants are altered by a wide range of pathophysiological conditions. Firstly, I Na is reduced by the impaired Na(+) channel function that arises clinically during heart failure, ischemia, tachycardia, and following treatment with class I antiarrhythmic drugs. Such reductions also arise as a consequence of mutations in SCN5A such as those occurring in Lenègre disease, Brugada syndrome (BrS), sick sinus syndrome, and atrial fibrillation (AF). Secondly, r a, may be increased due to gap junction decoupling following ischemia, ventricular hypertrophy, and heart failure, or as a result of mutations in CJA5 found in idiopathic AF and atrial standstill. Finally, either r a or c m could potentially be altered by fibrotic change through the resultant decoupling of myocyte-myocyte connections and coupling of myocytes with fibroblasts. Such changes are observed in myocardial infarction and cardiomyopathy or following mutations in MHC403 and SCN5A resulting in hypertrophic cardiomyopathy (HCM) or Lenègre disease, respectively. This review defines and quantifies the determinants of θ and summarizes experimental evidence that links changes in these determinants with reduced myocardial θ and arrhythmogenesis. It thereby identifies the diverse pathophysiological conditions in which abnormal θ may contribute to arrhythmia.
Keywords: arrhythmia; conduction velocity; fibrosis; gap junction; sodium channel.
Figures
References
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous